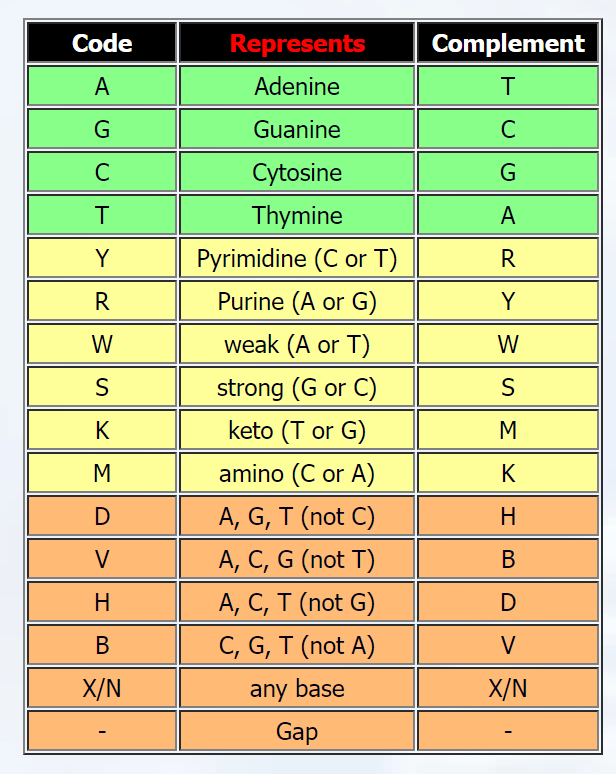
Nucleotide sequences (or DNA sequences) generally are comprised of 4 bases: ATGC which makes for a very nice, easy and efficient way of encoding this for machine learning purposes.
sequence = AAATGCC
ohe_sequence = [[1, 0, 0, 0], [1, 0, 0, 0], [1, 0, 0, 0], [0, 1, 0, 0], [0, 0, 1, 0], [0, 0, 0, 1], [0, 0, 0, 1]]
But when you take into account RNA sequences and mistakes that can sometimes occur in sequencing machines, the letters UYRWSKMDVHBXN are added... When one-hot encoding this you end up with a matrix of 17 rows of which the last 13 rows are generally all 0's.
This is very inefficient and does not confer the biological meaning that these extra (ambiguous) letters have.
For example:
- T and U are interchangeable
- Y represents there to be a C or T
- N and X represent there to be any of the 4 bases (ATGC)
And so I have made a dictionary that represents this biological meaning
nucleotide_dict = {'A': 'A', 'T':'T', 'U':'T', 'G':'G', 'C':'C', 'Y':['C', 'T'], 'R':['A', 'G'], 'W':['A', 'T'], 'S':['G', 'C'], 'K':['T', 'G'], 'M':['C', 'A'], 'D':['A', 'T', 'G'], 'V':['A', 'G', 'C'], 'H':['A', 'T', 'C'], 'B':['T', 'G', 'C'], 'X':['A', 'T', 'G', 'C'], 'N':['A', 'T', 'G', 'C']}
But I can't seem to figure out how to make an efficient one-hot-encoding script (or wether there is a way to do this with the scikit learn module) that utilizes this dictionary in order to get a result like this:
sequence = ANTUYCC
ohe_sequence = [[1, 0, 0, 0], [1, 1, 1, 1], [0, 1, 0, 0], [0, 1, 0, 0], [0, 1, 0, 1], [0, 0, 0, 1], [0, 0, 0, 1]]
# or even better:
ohe_sequence = [[1, 0, 0, 0], [0.25, 0.25, 0.25, 0.25], [0, 1, 0, 0], [0, 1, 0, 0], [0, 0.5, 0, 0.5], [0, 0, 0, 1], [0, 0, 0, 1]]
CodePudding user response:
This was fun! I think you can do this using a dictionary with the appropriate values. I added the scikit-learn class because you mentioned you were using that. See the transform below:
from sklearn.base import BaseEstimator, TransformerMixin
import numpy as np
nucleotide_dict = {
"A": [1, 0, 0, 0],
"G": [0, 1, 0, 0],
"C": [0, 0, 1, 0],
"T": [0, 0, 0, 1],
"U": [0, 0, 0, 1],
"Y": [0, 0, 1, 1],
"R": [1, 1, 0, 0],
"W": [1, 0, 0, 1],
"S": [0, 1, 1, 0],
"K": [0, 1, 0, 1],
"M": [1, 0, 1, 0],
"D": [1, 1, 0, 1],
"V": [1, 1, 1, 0],
"H": [1, 0, 1, 1],
"B": [0, 1, 1, 1],
"X": [1, 1, 1, 1],
"N": [1, 1, 1, 1],
"-": [0, 0, 0, 0],
}
class NucleotideEncoder(BaseEstimator, TransformerMixin):
def __init__(self, norm=True):
self.norm = norm
def fit(self, X, y=None):
return self
def transform(self, X, y=None):
f1 = lambda a: list(a)
f2 = lambda g: nucleotide_dict[g]
f3 = lambda c: list(map(f2, f1(c[0])))
f4 = lambda t: np.array(f3(t)) / np.sum(np.array(f3(t)), axis=1)[:, np.newaxis]
f = f3
if self.norm:
f = f4
return np.apply_along_axis(f, 1, X)
samples = np.array([["AAATGCC"], ["ANTUYCC"]])
print(NucleotideEncoder().fit_transform(samples))
Output:
[[[1. 0. 0. 0. ]
[1. 0. 0. 0. ]
[1. 0. 0. 0. ]
[0. 0. 0. 1. ]
[0. 1. 0. 0. ]
[0. 0. 1. 0. ]
[0. 0. 1. 0. ]]
[[1. 0. 0. 0. ]
[0.25 0.25 0.25 0.25]
[0. 0. 0. 1. ]
[0. 0. 0. 1. ]
[0. 0. 0.5 0.5 ]
[0. 0. 1. 0. ]
[0. 0. 1. 0. ]]]
CodePudding user response:
I like the latter approach, as it more closely corresponds to the real meaning: E.g. Y does not mean C and T, but one of C or T. If no further information is available, assuming equal probabilities (i.e. weights) seems reasonable. Of course this non-standard encoding needs to be reflected by the choice of loss function down the road.
To answer your question: You could precompute the mapping from the letters to the encoding and then create an encode function that takes a sequence as input and returns the encoded sequence as an (len(sequence), 4)-shaped np.array as follows:
import numpy as np
nucleotide_dict = {'A':'A', 'T':'T', 'U':'T', 'G':'G', 'C':'C', 'Y':['C', 'T'], 'R':['A', 'G'], 'W':['A', 'T'], 'S':['G', 'C'], 'K':['T', 'G'], 'M':['C', 'A'], 'D':['A', 'T', 'G'], 'V':['A', 'G', 'C'], 'H':['A', 'T', 'C'], 'B':['T', 'G', 'C'], 'X':['A', 'T', 'G', 'C'], 'N':['A', 'T', 'G', 'C']}
index_mapper = {'A': 0, 'T': 1, 'G': 2, 'C': 3}
mapper_dict = dict()
for k, v in nucleotide_dict.items():
encoding = np.zeros(4)
p = 1 / len(v)
encoding[[index_mapper[i] for i in v]] = p
mapper_dict[k] = encoding
def encode(sequence):
return np.array([mapper_dict[s] for s in sequence])
This seems to produce the desired result and is probably somewhat efficient.
An example:
print(encode('AYSDX'))
prints the following:
array([[1. , 0. , 0. , 0. ],
[0. , 0.5 , 0. , 0.5 ],
[0. , 0. , 0.5 , 0.5 ],
[0.33333333, 0.33333333, 0.33333333, 0. ],
[0.25 , 0.25 , 0.25 , 0.25 ]])
CodePudding user response:
Another version that allows for different length sequences:
import numpy as np
nucleotide_dict = {
"A": [1, 0, 0, 0],
"G": [0, 1, 0, 0],
"C": [0, 0, 1, 0],
"T": [0, 0, 0, 1],
"U": [0, 0, 0, 1],
"Y": [0, 0, 1, 1],
"R": [1, 1, 0, 0],
"W": [1, 0, 0, 1],
"S": [0, 1, 1, 0],
"K": [0, 1, 0, 1],
"M": [1, 0, 1, 0],
"D": [1, 1, 0, 1],
"V": [1, 1, 1, 0],
"H": [1, 0, 1, 1],
"B": [0, 1, 1, 1],
"X": [1, 1, 1, 1],
"N": [1, 1, 1, 1],
"-": [0, 0, 0, 0],
}
norm = True
samples = np.array(["AAATGCC", "ANTUYCC", "".join(list(nucleotide_dict.keys()))[:-1]])
def nucleotide_encode(samples, norm=True):
m = map(list, samples)
f1 = lambda x: np.array(list(map(nucleotide_dict.get, x)))
f = f1
if norm:
f = lambda x: np.nan_to_num(f1(x) / np.sum(f1(x), axis=1)[:, np.newaxis])
return list(map(f, m))
for i, j in zip(samples, nucleotide_encode(samples, norm=norm)):
print(i)
print(j)
Yields:
AAATGCC
[[1. 0. 0. 0.]
[1. 0. 0. 0.]
[1. 0. 0. 0.]
[0. 0. 0. 1.]
[0. 1. 0. 0.]
[0. 0. 1. 0.]
[0. 0. 1. 0.]]
ANTUYCC
[[1. 0. 0. 0. ]
[0.25 0.25 0.25 0.25]
[0. 0. 0. 1. ]
[0. 0. 0. 1. ]
[0. 0. 0.5 0.5 ]
[0. 0. 1. 0. ]
[0. 0. 1. 0. ]]
AGCTUYRWSKMDVHBXN
[[1. 0. 0. 0. ]
[0. 1. 0. 0. ]
[0. 0. 1. 0. ]
[0. 0. 0. 1. ]
[0. 0. 0. 1. ]
[0. 0. 0.5 0.5 ]
[0.5 0.5 0. 0. ]
[0.5 0. 0. 0.5 ]
[0. 0.5 0.5 0. ]
[0. 0.5 0. 0.5 ]
[0.5 0. 0.5 0. ]
[0.33333333 0.33333333 0. 0.33333333]
[0.33333333 0.33333333 0.33333333 0. ]
[0.33333333 0. 0.33333333 0.33333333]
[0. 0.33333333 0.33333333 0.33333333]
[0.25 0.25 0.25 0.25 ]
[0.25 0.25 0.25 0.25 ]]