I have a GRanges object:
library(GenomicRanges)
gr <- new("GRanges",
seqnames = new("Rle",
values = structure(1L, .Label = "chr8", class = "factor"),
lengths = 8L, elementMetadata = NULL, metadata = list()
),
ranges = new("IRanges",
start = c(
1L, 1L, 3L, 2L, 51L, 51L,
55L, 42L
), width = c(50L, 36L, 27L, 24L, 28L, 25L, 24L, 19L), NAMES = NULL, elementType = "ANY", elementMetadata = NULL,
metadata = list()
), strand = new("Rle",
values = structure(3L, .Label = c(
" ",
"-", "*"
), class = "factor"), lengths = 8L, elementMetadata = NULL,
metadata = list()
), seqinfo = new("Seqinfo",
seqnames = "chr8",
seqlengths = NA_integer_, is_circular = NA, genome = NA_character_
),
elementMetadata = new("DataFrame",
rownames = NULL, nrows = 8L,
listData = list(abundance = c(
101, 9982, 59, 310, 109,
478, 89, 403
), seq = c(
"GCCCGGCGTCCGTGGCCTTTGATCAAGGGCTTCCCTTTGCGAAAGCGCAT",
"TTGAGTTTACAGACTTTCTGCAATTTGGACGCCTGT",
"TCTAGTGCGAATATGGCTGGCCGTGAT",
"GTAAGCTGGCGGCCAGAATCCGGG",
"CGGCGCGGCAAGAACAACACGCGGTTCG",
"CCTGCCATCTGGCGAGTTCGGGACA",
"ACTTCGTGTGCGTTGGTTACTGAT",
"GCAAACGATGGATACAAGC"
)),
elementType = "ANY", elementMetadata = NULL,
metadata = list()
), elementType = "ANY", metadata = list()
)
It look like:
GRanges object with 8 ranges and 2 metadata columns:
seqnames ranges strand | abundance seq
<Rle> <IRanges> <Rle> | <numeric> <character>
[1] chr8 1-50 * | 101 GCCCGGCGTCCGTGGCCTTT..
[2] chr8 1-36 * | 9982 TTGAGTTTACAGACTTTCTG..
[3] chr8 3-29 * | 59 TCTAGTGCGAATATGGCTGG..
[4] chr8 2-25 * | 310 GTAAGCTGGCGGCCAGAATC..
[5] chr8 51-78 * | 109 CGGCGCGGCAAGAACAACAC..
[6] chr8 51-75 * | 478 CCTGCCATCTGGCGAGTTCG..
[7] chr8 55-78 * | 89 ACTTCGTGTGCGTTGGTTAC..
[8] chr8 42-60 * | 403 GCAAACGATGGATACAAGC
-------
seqinfo: 1 sequence from an unspecified genome; no seqlengths
Plot I can make
library(ggbio)
autoplot(gr, aes(fill = abundance))
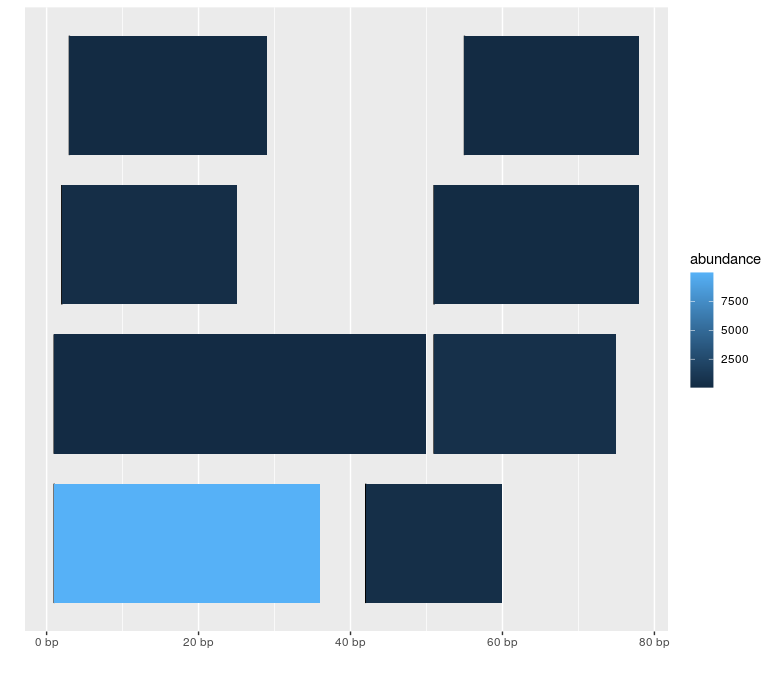
Question
How can I display the nucleotide sequences instead of the rectangles, with each nucleotide coloured differently?
CodePudding user response:
I personally find ggbio cumbersome to plot with, so my suggestion is to convert basepair-wise observations to a dataframe and then plot that.
library(GenomicRanges)
library(ggplot2)
# gr <- new("GRanges", ...) # omitted for brevity
# Make row for every letter
df <- mapply(function(s, e, seq, i) {
data.frame(
pos = seq(s, e, by = 1L),
seq = seq,
id = i
)
}, s = start(gr), e = end(gr), seq = strsplit(gr$seq, ""), i = seq_along(gr),
SIMPLIFY = FALSE)
# Combine different granges
df <- do.call(rbind, df)
# Determine y-position in plot
offset <- disjointBins(gr)
df$offset <- offset[df$id]
# Plot
ggplot(df, aes(pos, offset, fill = seq))
geom_tile(height = 0.8)
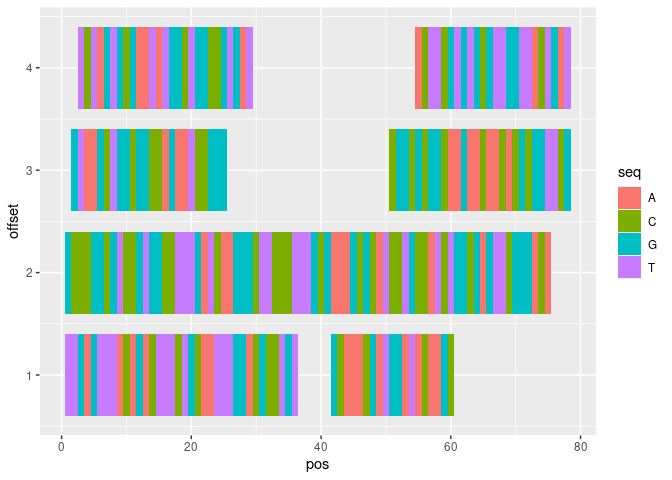
Created on 2021-10-11 by the reprex package (v2.0.0)