StackExchange.
I'm trying to build a phylogenetic tree for >100 individuals, using R.
However, while tutorials for packages like APE and ggtree readily show how to do this using raw DNA or pre-sorted groupings (e.g. (((A,B),(C,D)),E)), my data takes the form of a list of machine-numbered tags assembled from all over the genome.
For example:
sample <- c("A", "B", "C"...)
ID <- c("1 2 4 5", "2 4 5", "1 2 3 5"...)
df <- data.frame(sample, ID)
| sample | ID |
|---|---|
| A | 1 2 4 5 |
| B | 2 4 5 |
| C | 1 2 3 5 |
I'm struggling to figure out how (or even if it's possible) to build a phylogeny from this sort of arbitrary numbering. Is anybody familiar? (Rooted/unrooted/circular isn't particularly important)
Thanks for reading!
CodePudding user response:
A (phylogenetic) tree is just a hierarchical clustering of samples. The only thing needed is to define a dissimilarity measure between all sample pairs. In your case, you have sets of numbers for each row, and thus we can use Jaccard for this. For example, all elements of sample B are also found in sample A, so they should be put next to each other in the tree:
library(tidyverse)
library(proxy)
sample <- c("A", "B", "C")
ID <- c("1 2 4 5", "2 4 5", "1 2 3 5")
df <- data.frame(sample, ID)
df
#> sample ID
#> 1 A 1 2 4 5
#> 2 B 2 4 5
#> 3 C 1 2 3 5
distances <-
df %>%
separate_rows(ID) %>%
mutate(has_ID = 1) %>%
pivot_wider(names_from = ID, values_from = has_ID, values_fill = list(has_ID = 0)) %>%
column_to_rownames("sample") %>%
proxy::dist(by_rows = TRUE, method = "Jaccard")
distances
#> A B
#> B 0.25
#> C 0.40 0.60
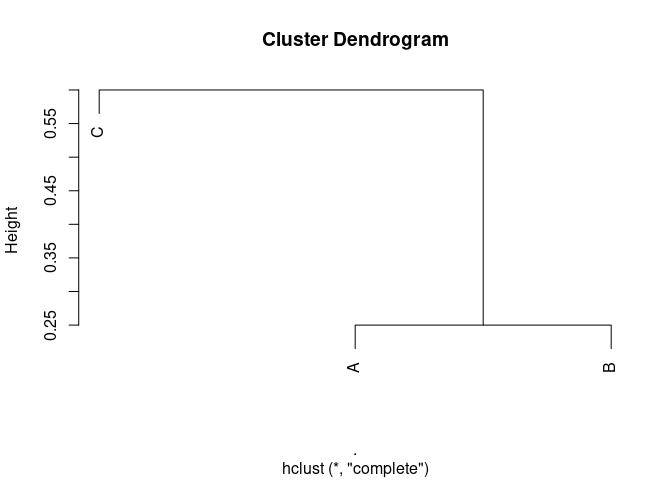
distances %>%
hclust() %>%
plot()
Created on 2022-05-13 by the reprex package (v2.0.0)