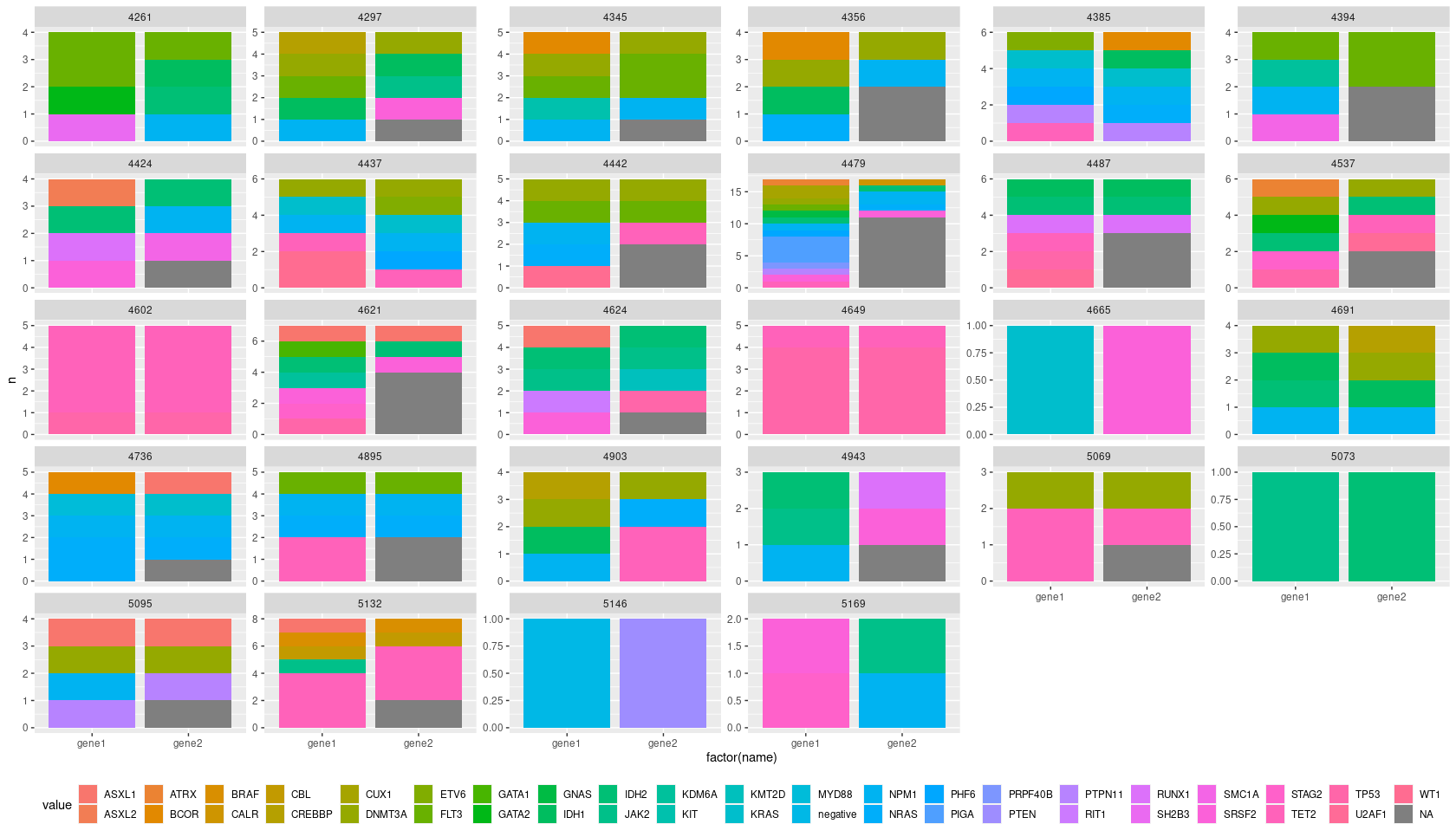
Dataframe df is shown:
ID gene1 gene2
4602 TET2 TET2
4602 TP53 TP53
4602 TET2 TET2
5095 ASXL1 ASXL1
5095 DNMT3A DNMT3A
5095 NPM1 <NA>
I have been trying to obtain a matching barplot displaying a count (bars) for both the column gene1 and gene2. the gene1 method is the standard, while gene2 is another mutation detector that should be compared to gene1. As you see, in sample 5095 only 2 mutations were detected, while the 3rd was not reproduced.
How do i make a barplot with two bars for each ID showing the count in gene1 and gene2?
here dput()
structure(list(ID = c(4602, 4602, 4602, 5095, 5095, 5095, 5095,
4649, 4649, 4649, 5069, 5069, 5069, 5146, 5132, 5132, 5132, 5132,
5132, 5132, 4297, 4297, 4297, 4297, 4297, 4345, 4345, 4345, 4345,
4345, 4356, 4356, 4356, 4356, 4385, 4385, 4385, 4385, 4385, 4385,
4437, 4437, 4437, 4437, 4437, 4437, 4442, 4442, 4442, 4442, 4442,
4479, 4479, 4479, 4479, 4479, 4479, 4479, 4479, 4479, 4479, 4479,
4479, 4479, 4479, 4479, 4487, 4487, 4487, 4487, 4487, 4487, 4537,
4537, 4537, 4537, 4537, 4537, 4621, 4621, 4621, 4621, 4621, 4621,
4621, 4624, 4624, 4624, 4624, 4624, 4665, 4736, 4736, 4736, 4736,
4736, 4895, 4895, 4895, 4895, 4895, 4903, 4903, 4903, 4903, 4691,
4691, 4691, 4691, 4261, 4261, 4261, 4261, 4394, 4394, 4394, 4394,
4424, 4424, 4424, 4424, 4943, 4943, 4943, 5073, 5169, 5169),
gene1 = c("TET2", "TP53", "TET2", "ASXL1", "DNMT3A", "NPM1",
"PTPN11", "TP53", "TP53", "TET2", "DNMT3A", "TET2", "TET2",
"negative", "JAK2", "ASXL1", "BRAF", "CBL", "TET2", "TET2",
"DNMT3A", "IDH1", "NPM1", "CREBBP", "FLT3", "DNMT3A", "FLT3",
"NPM1", "BCOR", "KIT", "DNMT3A", "IDH1", "NRAS", "BCOR",
"KRAS", "NPM1", "PTPN11", "ETV6", "PHF6", "TET2", "DNMT3A",
"KRAS", "NPM1", "WT1", "TET2", "WT1", "DNMT3A", "FLT3", "NPM1",
"NRAS", "WT1", "DNMT3A", "IDH2", "NPM1", "SRSF2", "ATRX",
"CUX1", "CUX1", "FLT3", "GNAS", "PHF6", "PIGA", "PIGA", "PRPF40B",
"PTPN11", "TET2", "IDH1", "IDH2", "RUNX1", "U2AF1", "TET2",
"TP53", "DNMT3A", "IDH2", "ATRX", "GATA2", "STAG2", "TP53",
"IDH2", "SRSF2", "ASXL1", "GATA1", "KDM6A", "STAG2", "TP53",
"IDH2", "JAK2", "SRSF2", "ASXL1", "RIT1", "KRAS", "NPM1",
"NRAS", "NRAS", "BCOR", "MYD88", "FLT3", "NPM1", "NRAS",
"TET2", "TET2", "DNMT3A", "IDH1", "NPM1", "CREBBP", "DNMT3A",
"IDH1", "IDH2", "NPM1", "FLT3", "FLT3", "GATA2", "SH2B3",
"FLT3", "NPM1", "KDM6A", "SMC1A", "IDH2", "SRSF2", "ASXL2",
"RUNX1", "IDH2", "JAK2", "NPM1", "JAK2", "SRSF2", "STAG2"
), gene2 = c("TET2", "TP53", "TET2", "ASXL1", "DNMT3A", NA,
"PTPN11", "TP53", "TP53", "TET2", "DNMT3A", NA, "TET2", "PTEN",
NA, NA, "BRAF", "CBL", "TET2", "TET2", "JAK2", "SRSF2", NA,
"DNMT3A", "IDH1", "NPM1", NA, "FLT3", "DNMT3A", "FLT3", "NPM1",
NA, NA, "DNMT3A", "IDH1", "NRAS", "BCOR", "KRAS", "NPM1",
"PTPN11", "ETV6", "PHF6", "TET2", "DNMT3A", "KRAS", "NPM1",
NA, "TET2", NA, "DNMT3A", "FLT3", "NPM1", "NRAS", NA, NA,
"IDH2", "NPM1", "SRSF2", NA, "CALR", NA, NA, NA, NA, NA,
NA, NA, NA, NA, "IDH1", "IDH2", "RUNX1", "U2AF1", "TET2",
NA, "DNMT3A", "IDH2", NA, NA, NA, NA, "IDH2", "SRSF2", "ASXL1",
NA, NA, "KMT2D", "TP53", "IDH2", "JAK2", "SRSF2", "ASXL1",
NA, "KRAS", "NPM1", "NRAS", "NRAS", NA, NA, "FLT3", "NPM1",
"NRAS", "TET2", "TET2", "DNMT3A", "IDH1", "NPM1", "CREBBP",
"DNMT3A", "IDH1", "IDH2", "NPM1", "FLT3", "FLT3", NA, NA,
"FLT3", "NPM1", NA, "SMC1A", "IDH2", "SRSF2", NA, "RUNX1",
"IDH2", "JAK2", "NPM1")), class = "data.frame", row.names = c(NA,
-127L))
CodePudding user response:
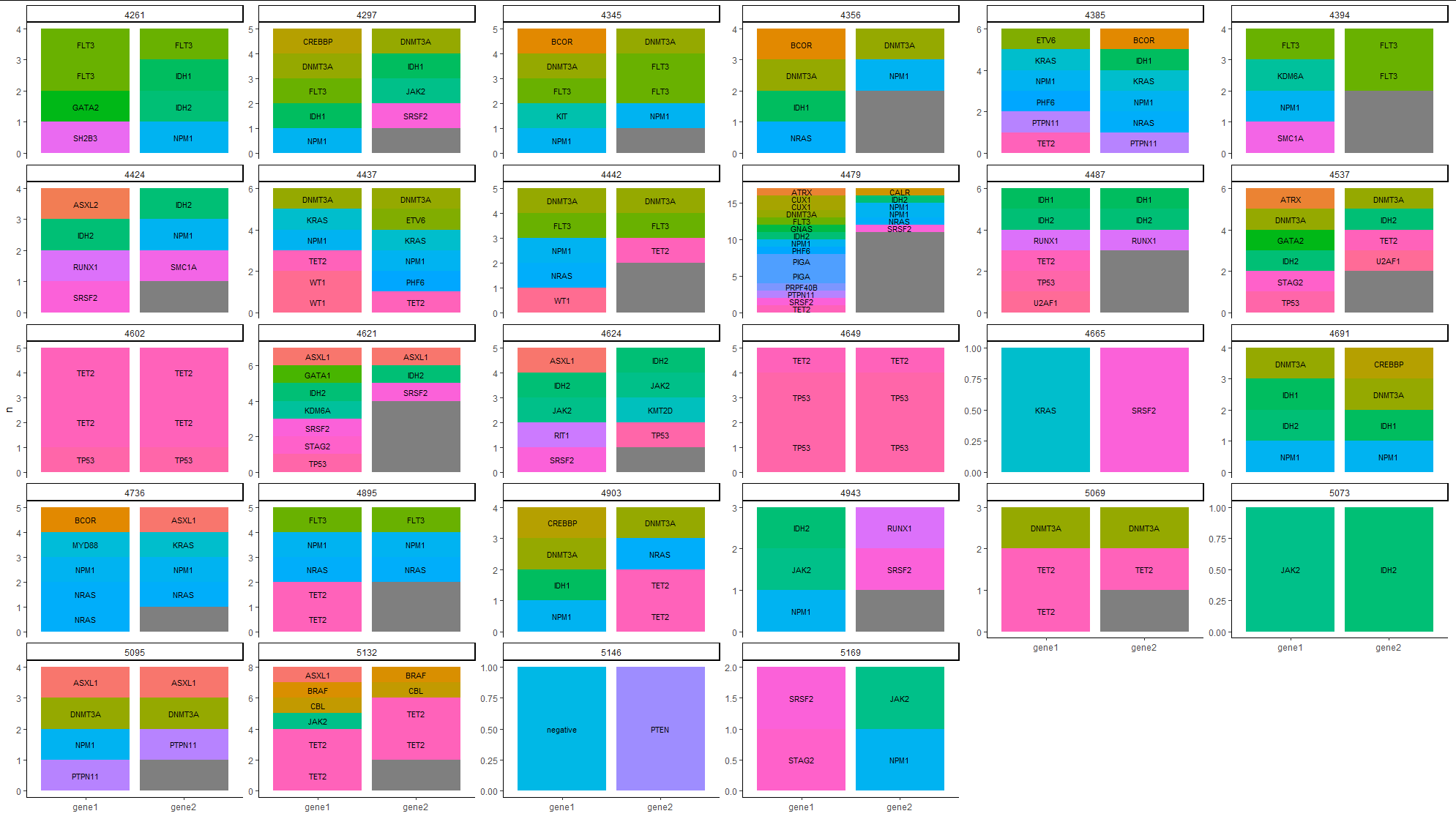
One more:
df %>%
group_by(ID) %>%
add_count(gene1, gene2) %>%
pivot_longer(
cols = contains("gene")
) %>%
ungroup() %>%
ggplot(aes(factor(name), n, fill=value, group=value, label=value))
geom_col()
facet_wrap(.~ID, scales = "free_y")
geom_text(size = 3, position = position_stack(vjust = 0.5))
theme_classic()
xlab("")
guides(fill=FALSE)
Last try, now it should work. Now we count before pivoting:
df %>%
group_by(ID) %>%
add_count(gene1, gene2) %>%
pivot_longer(
cols = contains("gene")
) %>%
ungroup() %>%
ggplot(aes(factor(name), n, fill=value, group=value))
geom_col()
facet_wrap(.~ID, scales = "free_y")
theme(legend.position = "bottom")
guides(fill=guide_legend(nrow=2))