I have a dataset called PimaDiabetes.
PimaDiabetes <- read.csv("PimaDiabetes.csv")
PimaDiabetes[2:8][PimaDiabetes[2:8]==0] <- NA
mean_1 = 40.5
mean_0 = 30.7
p.tib <- PimaDiabetes %>%
as_tibble()
Here is a snapshot of the data:
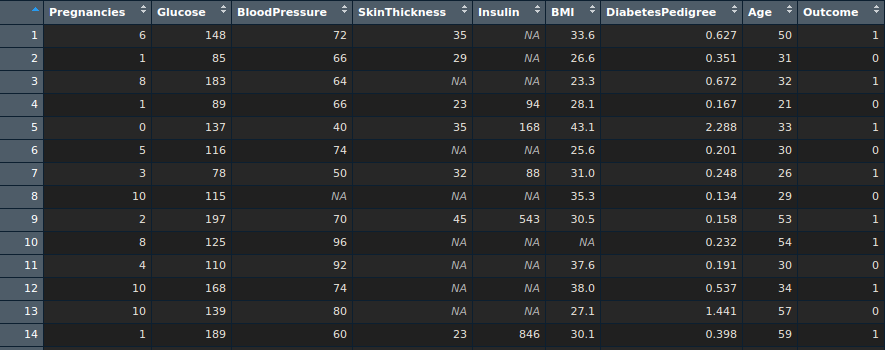
And the dataset can be pulled from here.
I'm trying to navigate the columns in such a way that I can group the dataset by Outcomes (so to select for Outcome 0 and 1), and impute a different value (the median of the respected groups) into columns depending on the outcomes.
So for instance, in the fifth column, Insulin, there are some NA values down the line where the Outcome is 1, and some where the Outcome is 0. I would like to place a value (40.5) into it when the value in a row is NA, and the Outcome is 1. Then I'd like to put the mean_2 into it when the value is NA, and the Outcome is 0.
I've gotten advice prior to this and tried:
p.tib %>%
mutate(
p.tib$Insulin = case_when((p.tib$Outcome == 0) & (is.na(p.tib$Insulin)) ~ IN_0,
(p.tib$Outcome == 1) & (is.na(p.tib$Insulin) ~ IN_1,
TRUE ~ p.tib$Insulin))
However it constantly yields the following error:
Error: unexpected '=' in "p.tib %>% mutate(p.tib$Insulin ="
Can I know where things are going wrong, please?
CodePudding user response:
Setup
It appears this dataset is also in the pdp package in R, called pima. The only major difference between the R package data and yours is that the pima dataset's Outcome variable is simply called "diabetes" instead and is labeled "pos" and "neg" instead of 0/1. I have loaded that package and the tidyverse to help.
#### Load Libraries ####
library(pdp)
library(tidyverse)
First I transformed the data into a tibble so it was easier for me to read.
#### Reformat Data ####
p.tib <- pima %>%
as_tibble()
Printing p.tib, we can see that the insulin variable has a lot of NA values in the first rows, which will be quicker to visualize later than some of the other variables that have missing data. Therefore, I used that instead of glucose, but the idea is the same.
# A tibble: 768 × 9
pregnant glucose press…¹ triceps insulin mass pedig…² age diabe…³
<dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <fct>
1 6 148 72 35 NA 33.6 0.627 50 pos
2 1 85 66 29 NA 26.6 0.351 31 neg
3 8 183 64 NA NA 23.3 0.672 32 pos
4 1 89 66 23 94 28.1 0.167 21 neg
5 0 137 40 35 168 43.1 2.29 33 pos
6 5 116 74 NA NA 25.6 0.201 30 neg
7 3 78 50 32 88 31 0.248 26 pos
8 10 115 NA NA NA 35.3 0.134 29 neg
9 2 197 70 45 543 30.5 0.158 53 pos
10 8 125 96 NA NA NA 0.232 54 pos
# … with 758 more rows, and abbreviated variable names ¹pressure,
# ²pedigree, ³diabetes
# ℹ Use `print(n = ...)` to see more rows
Finding the Mean
After glimpsing the data, I checked the mean for each group who did and didn't have diabetes by first grouping by diabetes with group_by, then collapsing the data frame into a summary of each group's mean, thus creating the mean_insulin variable (which you can see removes NA values to derive the mean):
#### Check Mean by Group ####
p.tib %>%
group_by(diabetes) %>%
summarise(mean_insulin = mean(insulin,
na.rm=T))
The values we should be imputing seem to be below. Here the groups are labeled as "neg" or 0 in your data, and "pos", or 1 in your data. You can convert these groups into those numbers if you want, but I left it as is so it was easier to read:
# A tibble: 2 × 2
diabetes mean_insulin
<fct> <dbl>
1 neg 130.
2 pos 207.
Mean Imputation
From there, we will use case_when as a vectorized ifelse statement. First, we use mutate to transform insulin. Then we use case_when by setting up three tests. First, if the group is negative and the value is NA, we turn it into the mean value of 130. If the group is positive for the same condition, we use 207. For all other values (the TRUE part), we just use the normal value of insulin. The & operator here just says "this transformation can only take place if both of these tests are true". What follows the ~ is the transformation to take place.
#### Impute Mean ####
p.tib %>%
mutate(
insulin = case_when(
(diabetes == "neg") & (is.na(insulin)) ~ 130,
(diabetes == "pos") & (is.na(insulin)) ~ 207,
TRUE ~ insulin
)
)
You will now notice that the first rows of insulin data are replaced with the mutation and the rest are left alone:
# A tibble: 768 × 9
pregnant glucose press…¹ triceps insulin mass pedig…² age diabe…³
<dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <fct>
1 6 148 72 35 207 33.6 0.627 50 pos
2 1 85 66 29 130 26.6 0.351 31 neg
3 8 183 64 NA 207 23.3 0.672 32 pos
4 1 89 66 23 94 28.1 0.167 21 neg
5 0 137 40 35 168 43.1 2.29 33 pos
6 5 116 74 NA 130 25.6 0.201 30 neg
7 3 78 50 32 88 31 0.248 26 pos
8 10 115 NA NA 130 35.3 0.134 29 neg
9 2 197 70 45 543 30.5 0.158 53 pos
10 8 125 96 NA 207 NA 0.232 54 pos
# … with 758 more rows, and abbreviated variable names ¹pressure,
# ²pedigree, ³diabetes
# ℹ Use `print(n = ...)` to see more rows