I have two datasets:
one is actual count and other one is predicted counts. I want to do a pearson correlation between them.
My actual count data look like this:
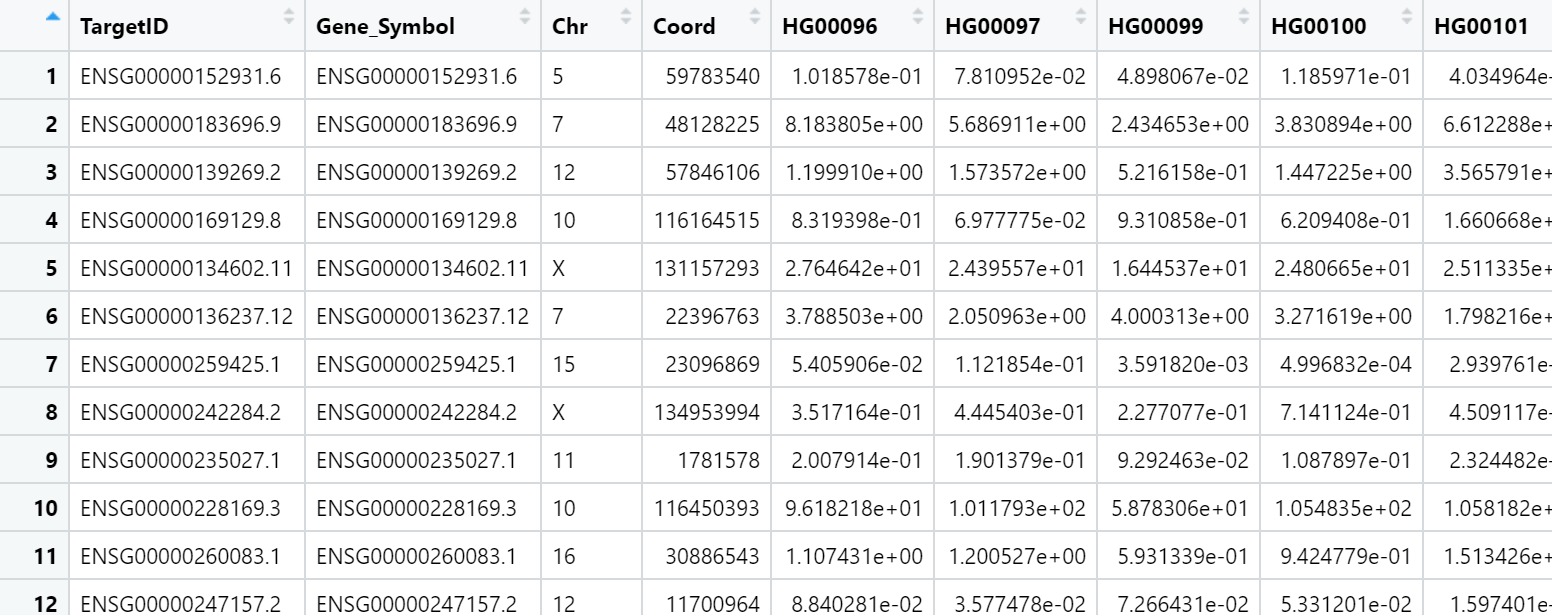 My predicted counts data look like this:
My predicted counts data look like this:
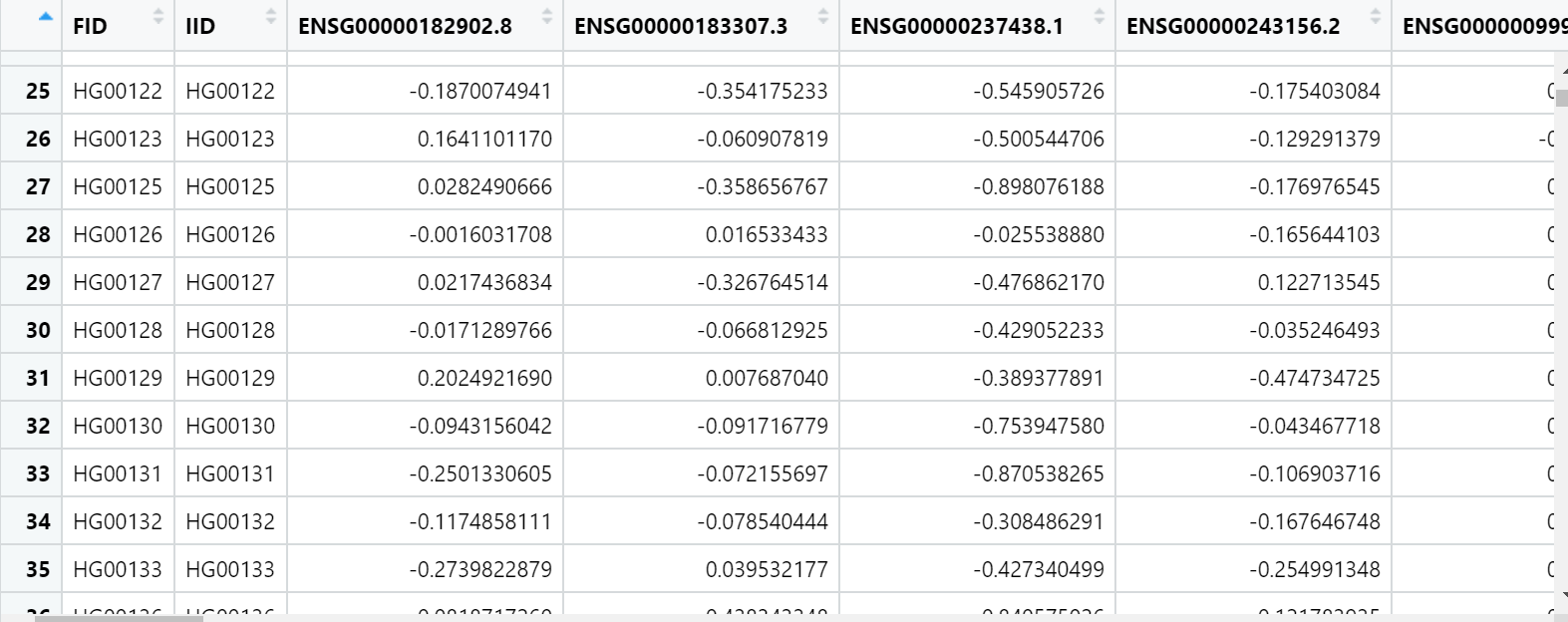
I want to do pearson correlation for these two datasets for each geneID.
I have written this code:
install.packages("Rcpp")
library(Rcpp)
library("reshape2")
library("ggplot2")
# import in the actual expression values and the gene predicted values
act_cts <- read.delim("GVDS_normalized_counts_2021v1.txt", header = TRUE, sep="\t")
## fix the column names
colnames(act_cts)[1]<-"gene"
colnames(act_cts)<- substr(colnames(act_cts), 1, 7)
pred_cts<-read.delim("GVDS_PrediXcan_Test_2021v1.txt", header=TRUE, sep="\t")
colnames(pred_cts)<-substr(colnames(pred_cts), 1, 15)
## melt the predict counts, so the columns change to row entries FID, IID, gene
melt_pred_cts<-melt(pred_cts, id.vars=c("FID","IID"), variable.name="gene", value.name = "gene_exp")
## melts the actual counts, so it can be easily joined to the final prediction
melt_act_cts<-melt(act_cts, id.vars="gene", variable.name="IID", value.name = "act_gene_exp")
final_cts<-merge(melt_pred_cts,melt_act_cts)
## this takes a minute/ several minutes to run because it is joining on both gene and IID
# runs the Pearson correlation for each gene
all_genes<-unique(final_cts$gene)
pear_cor_all_df<- data.frame(gene=character(), pear_coeff=double())
## runs the correlation
for(g in all_genes)
{
wrk_cts_all<-final_cts[which(final_cts$gene==g),]
# temp working df for each gene
pear_coef_all<-cor(wrk_cts_all$gene_exp, wrk_cts_all$act_gene_exp, method="pearson")
# runs the correlation for each gene between gene_exp and act_gene_exp
new_row_all<-c(g, pear_coef_all)
pear_cor_all_df<-rbind(pear_cor_all_df, new_row_all)
#saves this to the df
}
But its not giving me the correct results.
This is data for act_count:
dput(act_counts[1:10, 1:10])
structure(list(gene = c("ENSG00000152931.6", "ENSG00000183696.9",
"ENSG00000139269.2", "ENSG00000169129.8", "ENSG00000134602.11",
"ENSG00000136237.12", "ENSG00000259425.1", "ENSG00000242284.2",
"ENSG00000235027.1", "ENSG00000228169.3"), Gene_Sy = c("ENSG00000152931.6",
"ENSG00000183696.9", "ENSG00000139269.2", "ENSG00000169129.8",
"ENSG00000134602.11", "ENSG00000136237.12", "ENSG00000259425.1",
"ENSG00000242284.2", "ENSG00000235027.1", "ENSG00000228169.3"
), Chr = c("5", "7", "12", "10", "X", "7", "15", "X", "11", "10"
), Coord = c(59783540, 48128225, 57846106, 116164515, 131157293,
22396763, 23096869, 134953994, 1781578, 116450393), HG00096 = c(0.101857770468582,
8.1838049456063, 1.19991028786682, 0.831939826228749, 27.6464223725999,
3.78850273139249, 0.0540590649819536, 0.351716382898523, 0.200791414339667,
96.1821778045089), HG00097 = c(0.0781095249582053, 5.68691050653862,
1.57357169691446, 0.0697777450667378, 24.3955715036476, 2.05096276937706,
0.112185357489692, 0.444540251941709, 0.190137938062251, 101.17926156721
), HG00099 = c(0.0489806714207954, 2.43465332606958, 0.521615781673147,
0.93108575037257, 16.4453735152148, 4.00031300285966, 0.00359181983091798,
0.227707651999832, 0.0929246302159905, 58.7830634918037), HG00100 = c(0.118597118618172,
3.83089421985197, 1.44722544015787, 0.620940765480242, 24.8066495438254,
3.27161920134705, 0.00049968321150251, 0.714112406249513, 0.108789749488722,
105.483527339859), HG00101 = c(0.00403496367614745, 6.61228835251498,
3.56579072437701, 1.66066836204679, 25.1133488775017, 1.79821591847768,
0.0293976115522442, 0.450911709524112, 0.23244822901371, 105.818192023699
), HG00102 = c(0.0109253485646219, 4.70964559086586, 1.98268073472144,
0.570481056180073, 19.2339882617972, 1.51668840574531, 0.0312661751488703,
0.491437808951175, 0.250905117203001, 136.140843495464)), row.names = c(NA,
-10L), class = c("tbl_df", "tbl", "data.frame"))
This is prd_counts:
dput(prd_counts[1:10, 1:10])
structure(list(FID = c("HG00096", "HG00097", "HG00099", "HG00100",
"HG00101", "HG00102", "HG00103", "HG00105", "HG00106", "HG00107"
), IID = c("HG00096", "HG00097", "HG00099", "HG00100", "HG00101",
"HG00102", "HG00103", "HG00105", "HG00106", "HG00107"), ENSG00000182902.8 = c(0.0223611610092831,
0.0385031316687293, -0.0682504384265577, 0.00018098416274239,
-0.045492721345375, -0.10473163051734, -0.0215970711860838, 0.060455638944161,
-0.00889260689717109, -0.102096211855105), ENSG00000183307.3 = c(0.129041336028238,
-0.13226906002202, 0.005409246530295, -0.0539556427088601, -0.00699884042001628,
-0.204743560777908, -0.0534359750800079, -0.235648260835705,
-0.10230402771496, -0.0914043464852205), ENSG00000237438.1 = c(-0.758838434524167,
-0.579236418964912, -0.695762357174973, -0.368416879945024, -0.339555280234214,
-0.809438763600528, -0.359798980325098, -0.417769387016999, -0.724636782037491,
-0.309671271758401), ENSG00000243156.2 = c(-0.58456094489168,
0.105851861253113, -0.275061563982305, -0.0406543077034047, -0.522672785138957,
-0.126100301787985, -0.288382571274346, -0.354309857822533, -0.314842662063296,
-0.141401921597711), ENSG00000099968.13 = c(0.135357355615122,
0.157616292043257, 0.180059097593111, 0.250009792099489, 0.170653230854707,
0.316157576642492, 0.314671674077333, 0.224102148083679, 0.232969333848649,
0.14963210689311), ENSG00000069998.8 = c(-0.0346986034383362,
-0.0173493017191681, 0, -0.0173493017191681, -0.645266014640116,
-0.0346986034383362, -0.0173493017191681, -0.0173493017191681,
-0.0346986034383362, 0), ENSG00000184979.8 = c(-0.160573318589815,
0.54683218159596, 0.3503062647549, 0.653899917577768, 0.321280544783323,
0.653727041876318, 0.822864620159811, 1.03780221621802, -0.195295753744408,
-0.228590172992798), ENSG00000070413.12 = c(0.775225873145799,
0.602092262450708, 1.0198591935485, 0.65587457098494, 0.306445027670957,
0.581202299884586, 0.836112660742631, 0.559373823767867, 0.46977171007116,
0.84426113999649)), row.names = c(NA, -10L), class = c("tbl_df",
"tbl", "data.frame"))
CodePudding user response:
The provided test samples will not work because there are no genes in common between act_counts and prd_counts. I took the liberty of fixing that by reassigning column names:
library(dplyr)
library(tidyr)
## the line below fixes the problem with test samples
colnames(prd_counts)[3:10] <- act_counts$gene[1:8]
acts <- pivot_longer(act_counts,
cols = starts_with("HG"),
names_to = "FID",
values_to = "Actual")
prds <- pivot_longer(prd_counts,
cols = starts_with("ENSG"),
names_to = "gene",
values_to = "Predicted")
inner_join(acts, prds,
by = c("gene", "FID")) |>
select(gene, FID, Actual, Predicted) |>
group_by(gene) |>
summarize(rho = cor(Actual, Predicted))
##> # A tibble: 8 × 2
##> gene rho
##> <chr> <dbl>
##> 1 ENSG00000134602.11 -0.445
##> 2 ENSG00000136237.12 0.446
##> 3 ENSG00000139269.2 0.543
##> 4 ENSG00000152931.6 0.770
##> 5 ENSG00000169129.8 -0.802
##> 6 ENSG00000183696.9 0.405
##> 7 ENSG00000242284.2 -0.503
##> 8 ENSG00000259425.1 -0.110