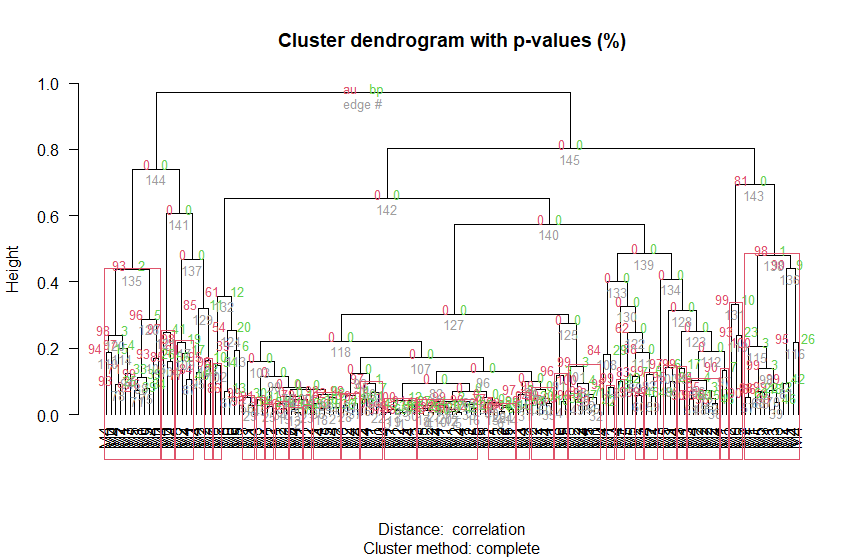
This is what I'm doing
mat <- read.table("Model_pvclust/Model18_FAB_M5_vs_M0_MAP_TF.txt",sep = "\t",strip.white = FALSE,check.names = FALSE,header=TRUE,row.names=1)
drop <- c("gene","baseMean","log2FoldChange","lfcSE","stat","pvalue","padj","UP_DOWN")
d1 <- select(mat, -one_of(drop))
####### Read the metadata
metadata <-read.table("Model_hmap_meta/FAB_table.txt",sep = "\t",strip.white = FALSE,check.names = FALSE,header=TRUE,row.names=1)
head(metadata)
head(metadata)
prior_malignancy FAB Risk_Cyto
TCGA-AB-2856 no M4 Intermediate
TCGA-AB-2849 no M0 Poor
TCGA-AB-2971 no M4 Intermediate
TCGA-AB-2930 no M2 Intermediate
TCGA-AB-2891 no M1 Poor
TCGA-AB-2872 no M3 Good
Plotting the cluster
pvc <- pvclust( data = d1 , method.dist = "correlation", method.hclust = "complete",parallel = T)
plot(pvc,las=2,hang = -0.5)
pvrect(pvc, alpha = 0.9)
Image that i get is where My sample names are labelled. Instead of those sample names I would like to label them based on that FAB columns matching the sample name order.
My data files 
Updated answer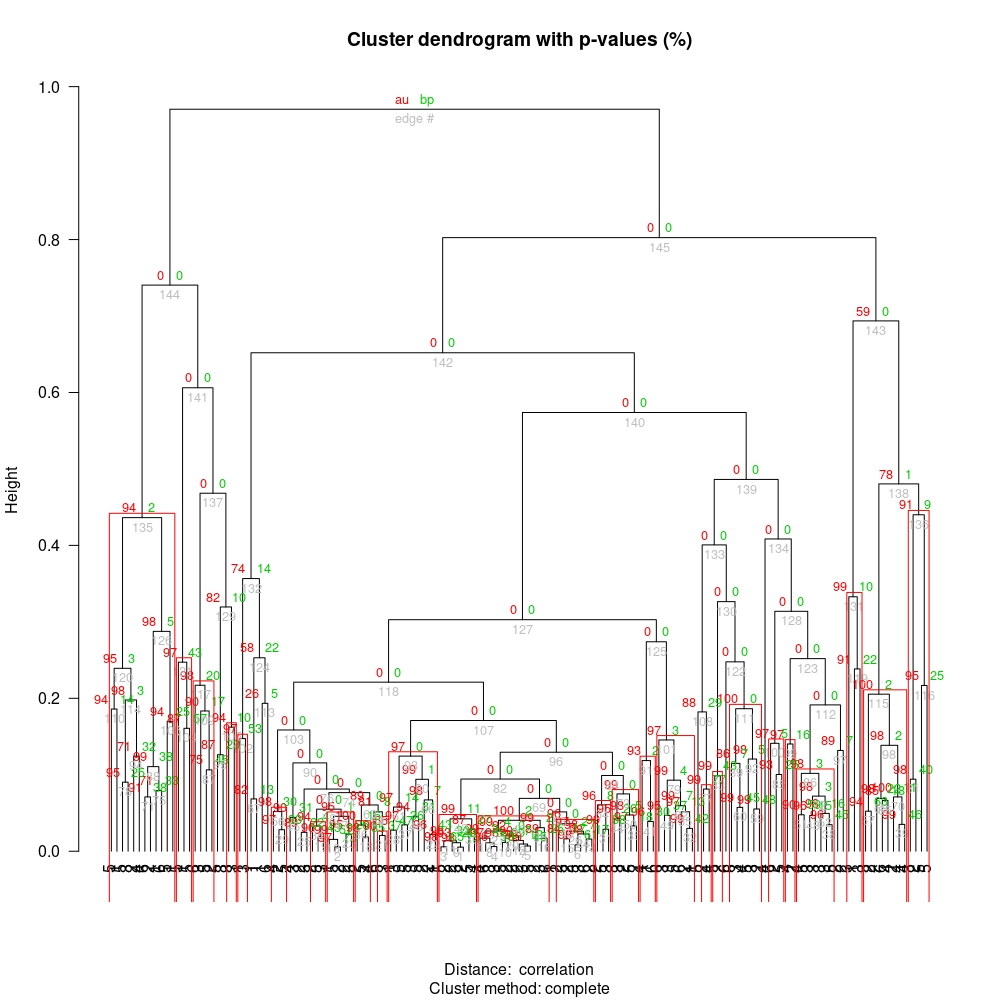
CodePudding user response:
You can replace the current labels using the dendextend::labels function.
library("dendextend")
labels(pvc$hclust) <- metadata$FAB
plot(pvc,las=2,hang = -0.5)
pvrect(pvc, alpha = 0.9)