I have data that look like this:
| Gene | HBEC-KT-01 | HBEC-KT-02 | HBEC-KT-03 | HBEC-KT-04 | HBEC-KT-05 | Primarycells-02 | Primarycells-03 | Primarycells-04 | Primarycells-05 |
|---|---|---|---|---|---|---|---|---|---|
| BPIFB1 | 15726000000 | 15294000000 | 15294000000 | 14741000000 | 22427000000 | 87308000000 | 2.00E 11 | 1.04E 11 | 1.51E 11 |
| LCN2 | 18040000000 | 26444000000 | 28869000000 | 30337000000 | 10966000000 | 62388000000 | 54007000000 | 56797000000 | 38414000000 |
| C3 | 2.52E 11 | 2.26E 11 | 1.80E 11 | 1.80E 11 | 1.78E 11 | 46480000000 | 1.16E 11 | 69398000000 | 78766000000 |
| MUC5AC | 15647000 | 8353200 | 12617000 | 12221000 | 29908000 | 40893000000 | 79830000000 | 28130000000 | 69147000000 |
| MUC5B | 965190000 | 693910000 | 779970000 | 716110000 | 1479700000 | 38979000000 | 90175000000 | 41764000000 | 50535000000 |
| ANXA2 | 14705000000 | 18721000000 | 21592000000 | 18904000000 | 22657000000 | 28163000000 | 24282000000 | 21708000000 | 16528000000 |
I want to make a heatmap like the following using R. I am following a paper and they quoted "Heat maps were generated with the ‘pheatmap’ package76, where correlation clustering distance row was applied". Here is their heatmap.
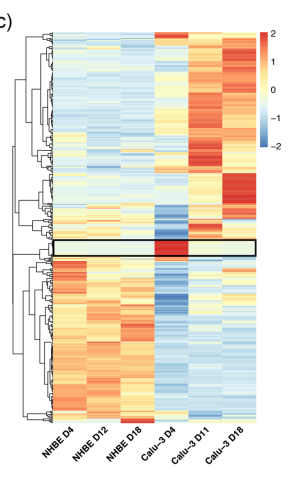
I want the same like this and I am trying to make one using R by following tutorials but I am new to R language and know nothing about R.
Here is my code.
df <- read.delim("R.txt", header=T, row.names="Gene")
df_matrix <- data.matrix(df)
pheatmap(df_matrix,
main = "Heatmap of Extracellular Genes",
color = colorRampPalette(rev(brewer.pal(n = 10, name = "RdYlBu")))(10),
cluster_cols = FALSE,
show_rownames = F,
fontsize_col = 10,
cellwidth = 40,
)
This is what I get.

When I try using clustering, I got the error.
pheatmap(
mat = df_matrix,
scale = "row",
cluster_column = F,
show_rownames = TRUE,
drop_levels = TRUE,
fontsize = 5,
clustering_method = "complete",
main = "Hierachical Cluster Analysis"
)
Error in hclust(d, method = method) :
NA/NaN/Inf in foreign function call (arg 10)
Can someone help me with the code?
CodePudding user response:
You can normalize the data using scale to archive a more uniform coloring. Here, the mean expression is set to 0 for each sample. Genes lower expressed than average have a negative z score:
library(tidyverse)
library(pheatmap)
data <- tribble(
~Gene, ~`HBEC-KT-01`, ~`HBEC-KT-02`, ~`HBEC-KT-03`, ~`HBEC-KT-04`, ~`HBEC-KT-05`, ~`Primarycells-03`, ~`Primarycells-04`, ~`Primarycells-05`,
"BPIFB1", 1.5726e 10, 1.5294e 10, 1.5294e 10, 1.4741e 10, 2.2427e 10, 2e 11, 1.04e 11, 1.51e 11,
"LCN2", 1.804e 10, 2.6444e 10, 2.8869e 10, 3.0337e 10, 1.0966e 10, 5.4007e 10, 5.6797e 10, 3.8414e 10,
"C3", 2.52e 11, 2.26e 11, 1.8e 11, 1.8e 11, 1.78e 11, 1.16e 11, 6.9398e 10, 7.8766e 10,
"MUC5AC", 15647000, 8353200, 12617000, 12221000, 29908000, 7.983e 10, 2.813e 10, 6.9147e 10,
"MUC5B", 965190000, 693910000, 779970000, 716110000, 1479700000, 9.0175e 10, 4.1764e 10, 5.0535e 10,
"ANXA2", 1.4705e 10, 1.8721e 10, 2.1592e 10, 1.8904e 10, 2.2657e 10, 2.4282e 10, 2.1708e 10, 1.6528e 10
)
data %>%
mutate(across(where(is.numeric), scale)) %>%
column_to_rownames("Gene") %>%
pheatmap(
scale = "row",
cluster_column = F,
show_rownames = FALSE,
show_colnames = TRUE,
treeheight_col = 0,
drop_levels = TRUE,
fontsize = 5,
clustering_method = "complete",
main = "Hierachical Cluster Analysis (z-score)",
)
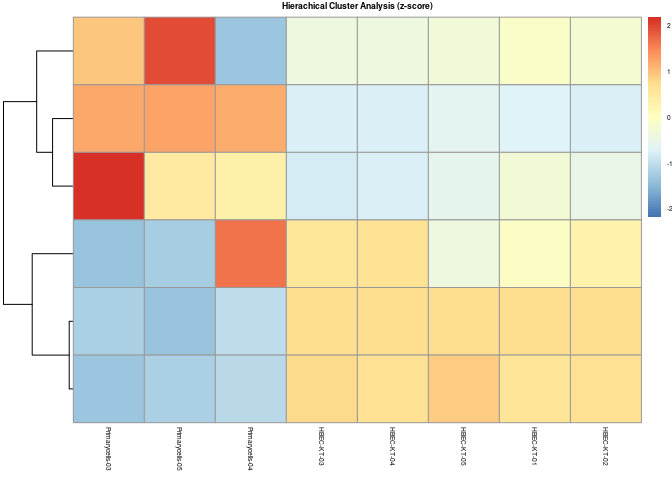
Created on 2021-09-26 by the reprex package (v2.0.1)