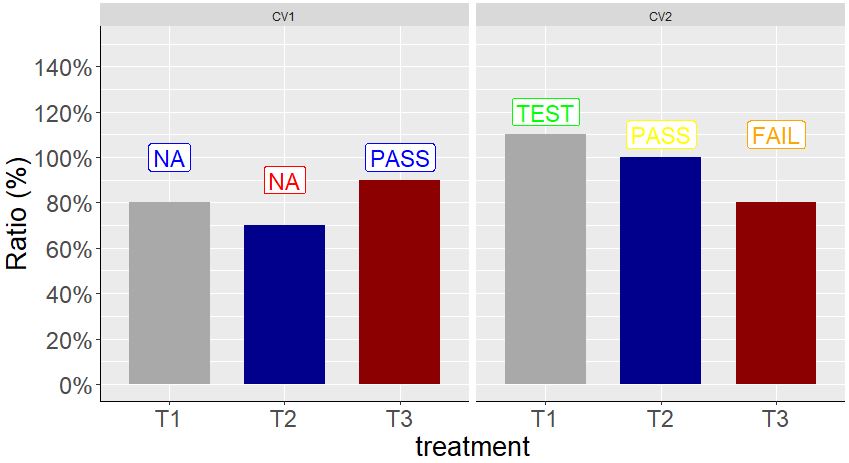
Here is a data
Genotype<- rep(c("CV1","CV2"),each=3)
treatment<-rep(c("T1","T2","T3"),2)
RATIO<- c(0.8,0.7,0.9,1.1,1.0,0.8)
DataA<- data.frame(Genotype,treatment,RATIO)
and this is a graph using facet_wrap()
A_labels <- data.frame(Genotype=c("CV1", "CV1", "CV1"),
perc= c("NA", "NA","PASS"), treatment=c("T1", "T2", "T3"),
RATIO=c(1.0, 0.90, 1.0))
B_labels <- data.frame(Genotype=c("CV2", "CV2", "CV2"),
perc= c("TEST", "PASS","FAIL"), treatment=c("T1", "T2", "T3"),
RATIO=c(1.2, 1.1, 1.1))
ggplot(data=DataA, aes(x=treatment, y=RATIO, fill=treatment))
geom_bar(stat="identity",position="dodge", width = 0.7)
scale_fill_manual(values= c ("Dark gray","Dark blue", "Dark red"))
scale_y_continuous(breaks = seq(0,1.5,0.2), labels = scales::percent, limits = c(0,1.5))
geom_label(data=A_labels, aes(x=treatment, y=RATIO, label=perc, col=treatment), size=6, fill="white")
geom_label(data=B_labels, aes(x=treatment, y=RATIO, label=perc, col=treatment), size=6, fill="white")
scale_color_manual(values= c ("Blue","Red", "Blue"))
labs(x="treatment", y="Ratio (%)")
facet_wrap(~Genotype)
theme(axis.title = element_text (face = "plain", size = 20, color = "black"),
axis.text.x = element_text(size= 18),
axis.text.y = element_text(size= 18),
axis.line = element_line(size = 0.5, colour = "black"),
legend.position = 'none')
windows(width=9, height=5)
It was successful to place texts in each panel independently using facet_wrap(), but I don't know how to put different color per treatment in each panel independently. For example, in the right panel, I want to change text color as green, yellow and orange.
Due to the code scale_color_manual(values= c ("Blue","Red", "Blue")) text color per treatment is the same in both panels.
Could you let me know how to change text color per treatment in each panel independently?
Thanks,
CodePudding user response:
As you mentioned, you have only two levels. So I would do this a bit differently:
#Loading the data and creating the the labels
Genotype<- rep(c("CV1","CV2"),each=3)
treatment<-rep(c("T1","T2","T3"),2)
RATIO<- c(0.8,0.7,0.9,1.1,1.0,0.8)
DataA<- data.frame(Genotype,treatment,RATIO)
A_labels <- data.frame(Genotype=c("CV1", "CV1", "CV1"),
perc= c("NA", "NA","PASS"), treatment=c("T1", "T2", "T3"),
RATIO=c(1.0, 0.90, 1.0))
B_labels <- data.frame(Genotype=c("CV2", "CV2", "CV2"),
perc= c("TEST", "PASS","FAIL"), treatment=c("T1", "T2", "T3"),
RATIO=c(1.2, 1.1, 1.1))
Now, I will create two separate plots for two levels
ggplot(data=subset(DataA, Genotype=='CV1'), aes(x=treatment, y=RATIO, fill=treatment))
geom_bar(stat="identity",position="dodge", width = 0.7)
scale_fill_manual(values= c ("Dark gray","Dark blue", "Dark red"))
scale_y_continuous(breaks = seq(0,1.5,0.2), labels = scales::percent, limits = c(0,1.5))
geom_label(data=A_labels, aes(x=treatment, y=RATIO, label=perc, col=treatment), size=6, fill="white")
scale_color_manual(values= c ("Blue","Red", "Blue"))
facet_wrap(~Genotype)
labs(x="treatment", y="Ratio (%)")
theme(axis.title = element_text (face = "plain", size = 20, color = "black"),
axis.text.x = element_text(size= 18),
axis.text.y = element_text(size= 18),
axis.line = element_line(size = 0.5, colour = "black"),
legend.position = 'none')-> plot_cv1
ggplot(data=subset(DataA, Genotype=='CV2'), aes(x=treatment, y=RATIO, fill=treatment))
geom_bar(stat="identity",position="dodge", width = 0.7)
scale_fill_manual(values= c ("Dark gray","Dark blue", "Dark red"))
scale_y_continuous(breaks = seq(0,1.5,0.2), labels = scales::percent, limits = c(0,1.5))
geom_label(data=B_labels, aes(x=treatment, y=RATIO, label=perc, col=treatment), size=6, fill="white")
scale_color_manual(values= c ("Green","Yellow", "Orange"))
facet_wrap(~Genotype)
labs(x="treatment", y="Ratio (%)")
theme(axis.title = element_text (face = "plain", size = 20, color = "black"),
axis.text.x = element_text(size= 18),
axis.text.y = element_text(size= 18),
axis.line = element_line(size = 0.5, colour = "black"),
legend.position = 'none')-> plot_cv2
Now you may use, grid.arrange from gridExtra or ggarrange from ggpubr
library(gridExtra)
grid.arrange(plot_cv1, plot_cv2, nrow = 1)
#or
library(ggpubr)
ggarrange(plot_cv1, plot_cv2, nrow = 1)
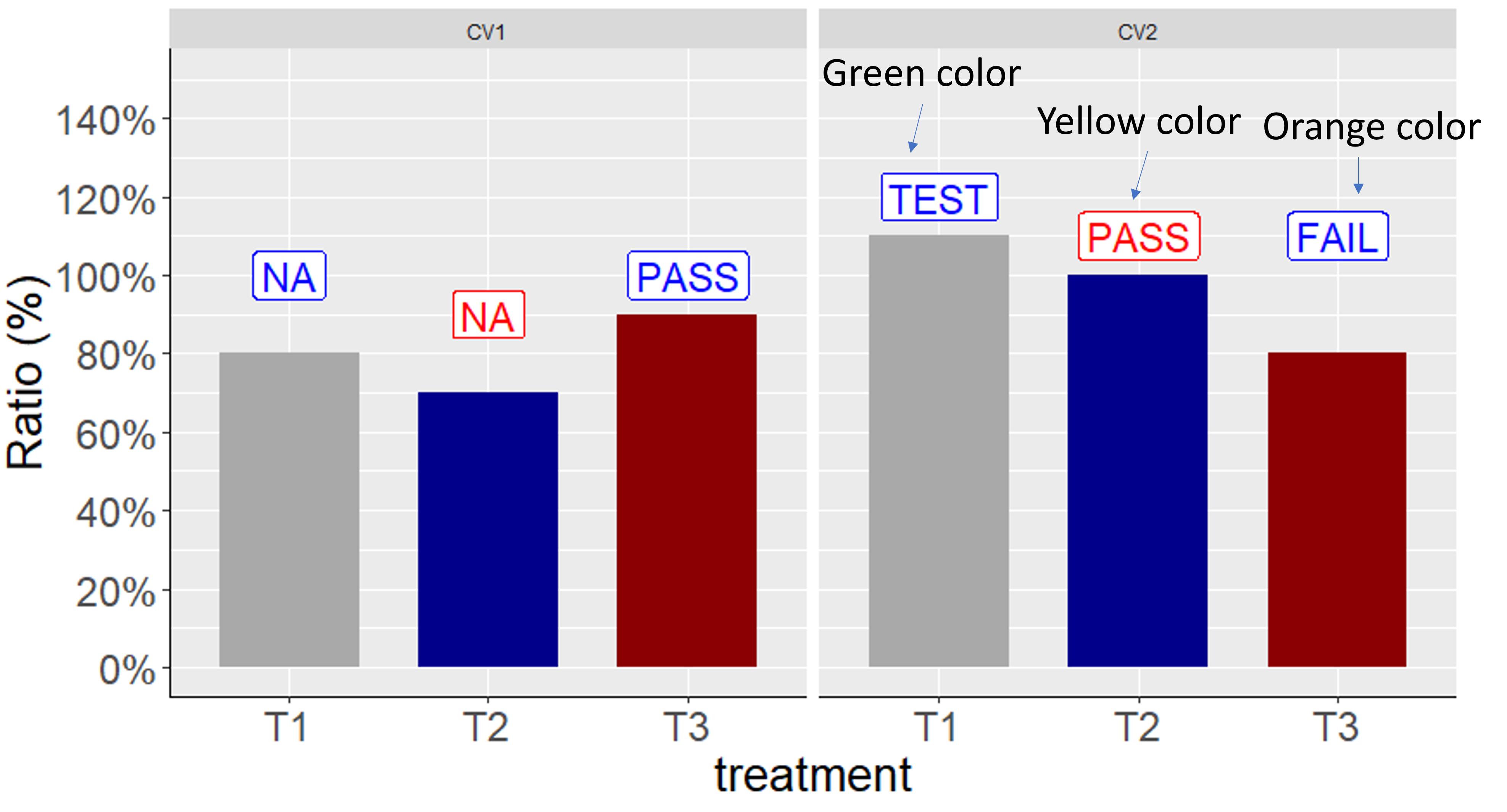
Both will have the same output:
CodePudding user response:
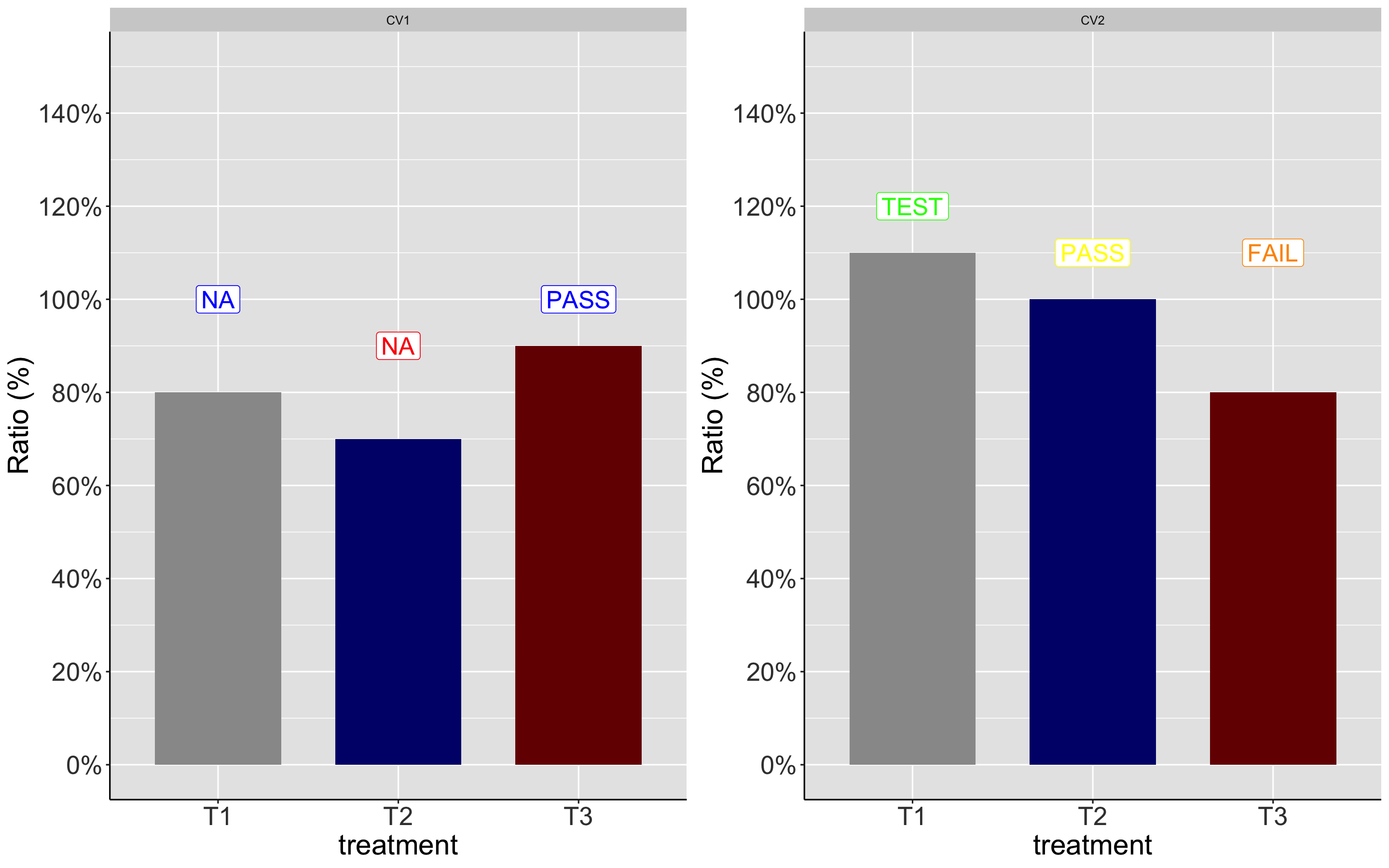
I found the way to solve this issue from the Ben's advice above. First we make one combination between 2 factors, and this combination will have 6 levels. (2 Genotype x 3 treatment). Then, in scale_color_manual, we can designate each color about the 6 levels!!
#Loading the data and creating the the labels
Genotype<- rep(c("CV1","CV2"),each=3)
treatment<-rep(c("T1","T2","T3"),2)
COMBI<- c("CV1T1","CV1T2","CV1T3","CV2T1","CV2T2","CV2T3")
RATIO<- c(0.8,0.7,0.9,1.1,1.0,0.8)
DataA<- data.frame(Genotype,treatment,COMBI, RATIO)
A_labels <- data.frame(Genotype=c("CV1", "CV1", "CV1"),
perc= c("NA", "NA","PASS"), treatment=c("T1", "T2", "T3"),
RATIO=c(1.0, 0.90, 1.0))
B_labels <- data.frame(Genotype=c("CV2", "CV2", "CV2"),
perc= c("TEST", "PASS","FAIL"), treatment=c("T1", "T2", "T3"),
RATIO=c(1.2, 1.1, 1.1))
#GGPLOT
ggplot(data=DataA, aes(x=treatment, y=RATIO, fill=treatment))
geom_bar(stat="identity",position="dodge", width = 0.7)
scale_fill_manual(values= c ("Dark gray","Dark blue", "Dark red"))
scale_y_continuous(breaks = seq(0,1.5,0.2), labels = scales::percent, limits = c(0,1.5))
geom_label(data=A_labels, aes(x=treatment, y=RATIO, label=perc, col=subset(DataA, Genotype=="CV1")$COMBI), size=6, fill="white")
geom_label(data=B_labels, aes(x=treatment, y=RATIO, label=perc, col=subset(DataA, Genotype=="CV2")$COMBI), size=6, fill="white")
scale_color_manual(values= c ("Blue","Red","Blue","Green","Yellow", "Orange"))
labs(x="treatment", y="Ratio (%)")
facet_wrap(~Genotype)
theme(axis.title = element_text (face = "plain", size = 20, color = "black"),
axis.text.x = element_text(size= 18),
axis.text.y = element_text(size= 18),
axis.line = element_line(size = 0.5, colour = "black"),
legend.position = 'none')
windows(width=9, height=5)