Rookie, I was born a letter when using hisat2 comparing to the genome sequence appeared many problems, the following simple said I tread pit, 1, personal feel, unless you want to build contains exons and splice site index, if just compare to the genome, can be directly downloaded from hisat2 website reference index in the genome, in a folder after download (human is hg19 if download hg19 mm10) in mice, which are genome. 123,,,. Ht2 file 2, when using hisat2 comparison, the best index, x.h t2, fq. Gz file, advance deposit. Sam into the same folder, or may be wrong, and I don't know why because I very vegetables 3, before use hisat2 must hisat2 - h see the command, or you'll, pound-foolish! Hisat2 than general parameters of the following Hisat2 - p - dta - 8 x hg19/genome - 1 sample_1. Fq. Gz - 2 sample_2. Fq. Gz - S sample. Sam -p 8 general how many nuclear to run, this watch your computer memory, 16 g I choose eight nuclear slightly point card, - dta is report, - x is your identity, hg19 is your deposit index directory, the genome is your prefix index file! Interrupt at this time, if you don't have to see hisat2 requirement, you may be behind the hg19 directly to the folder in - x, or will be one of the genome. X.h t2 using regular expressions genome. * held behind - x, or you could use the cat> the whole into a genome, these will cause hisat2 don't know you of the index file, see hisat2 request, you will understand, the somebody else just want prefix of the index, so you need to put hg19/genome - x can back! - 1-2 behind fastq file must be written on the path, or like I said to put them into the same folder, folder named I like to use alignedThis path can't go wrong and hisat2 won't convulsions error! 4, if your fastq file a lot, you can use for the terminal... do... Done to write a simple loop for I in xx yy 'do' seq hisat2-8 - x hg19/p genome (all of your index file prefix) - 1 sample ${I} 1. Fq. Gz - 2 sample ${I} 2. Fq. Gz - S sample ${I}. Sam If you want to write a sh in vim, pay attention to don't forget to configure the PATH 5, not must ask more and more don't behind closed doors, or a waste of time! Wish everyone born letter as a master! Finally append a train diagrams (my Ubuntu is so lovely, and the penguin although this is the label but I still want to use centos haha)
CodePudding user response:
Hisat2 I used for the construction of a genome index file, has been running for two days and two nights, but still didn't finish, I was thinking, the need for such a long time?
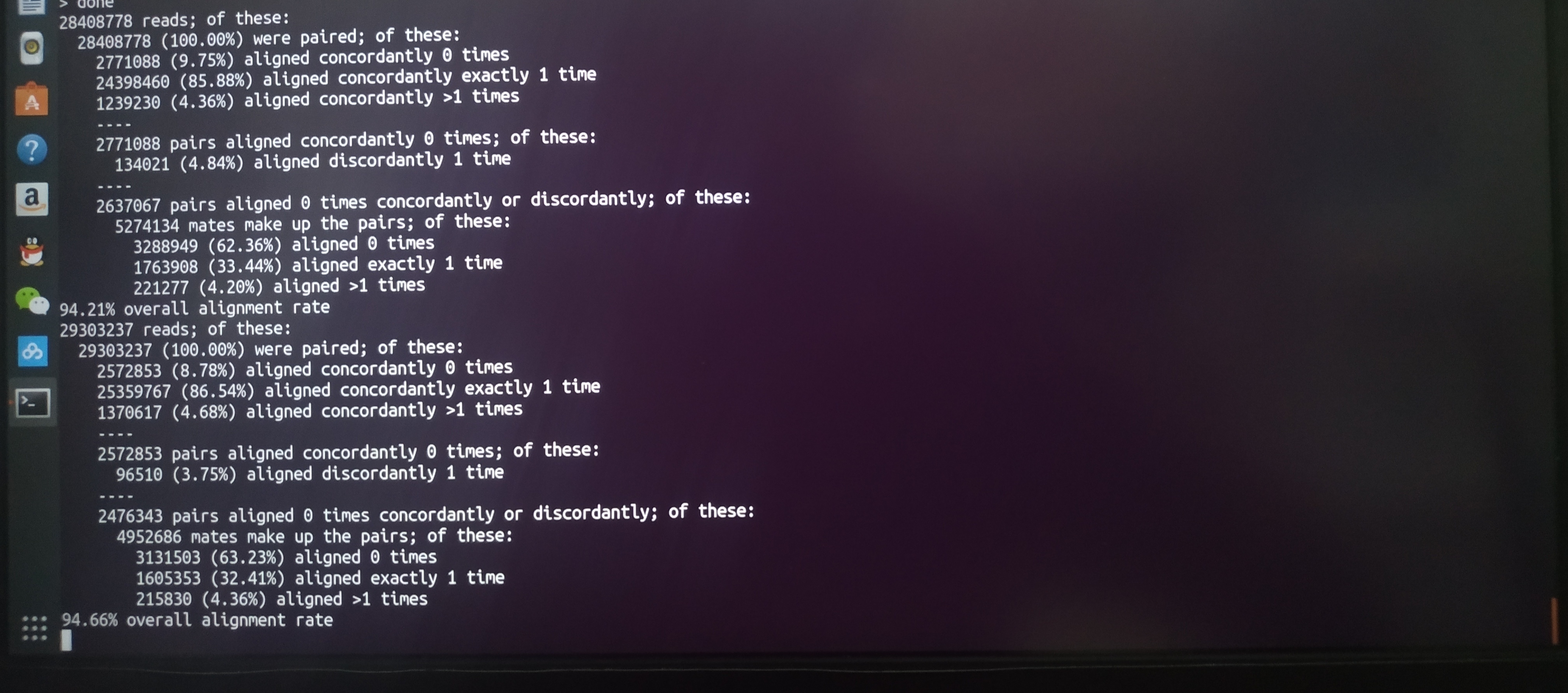 although this is the label but I still want to use centos haha)
although this is the label but I still want to use centos haha)