I really hope you have time to help out here. So, I'm trying to do a plot where I want to remove the default legend, simply because it doesn't make any sense because it is based on size that is from 0-1 (I tried to use legend.position = "none" which worked) but then I wanted to add a new legend that I make based on my plot, so that I have three options: small dot (log fold < 2), blue dot (p < 0.001 and log fold < 2), and red dot (p < 0.001 and log fold > 2) according to my graph. But I can't get to remove the default legend and still add a new legend?!
Thanks a lot in advance!
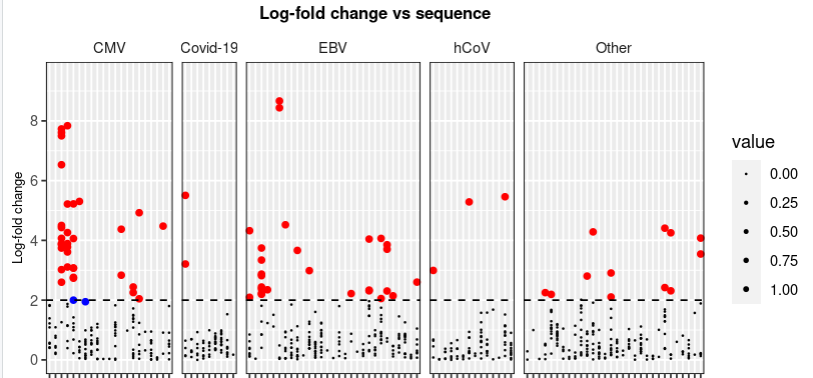
Below is my code to plot the graph..
maximum_y <- my_data_clean_aug %>%
pull(log_fold_change) %>%
max() %>%
round() 0.5
#Determining the numbers of sequences per virus strain (Origin) and setting a threshold.
threshold <- my_data_clean_aug %>%
count(Origin) %>%
filter(n > 50) %>%
count() %>%
pull()
#Pooling all groups of vira with less than 50 hits into HHV or Others
my_data_clean_aug_pooling <- my_data_clean_aug %>%
mutate(Origin = as.factor(Origin)) %>%
mutate(newID = fct_lump(Origin, threshold)) %>%
mutate(value = case_when(log_fold_change <= 2 ~ 0,
0.001 < p & log_fold_change >= 2 ~ 0,
0.001 >= p & log_fold_change >= 2 ~ 1))
pointsofinterest <- my_data_clean_aug_pooling %>%
filter(0.001 >= p & log_fold_change >= 2)
pointswithpsig <- my_data_clean_aug_pooling %>%
filter(0.001 >= p & log_fold_change < 2)
my_data_clean_aug_pooling %>%
ggplot(aes(x = Peptide,
y = log_fold_change))
facet_grid(.~newID,
scales = "free_x",
space = "free")
geom_point(aes_string(size = "value"))
geom_point(data = pointsofinterest,
color = "red")
geom_point(data = pointswithpsig,
color = "blue")
geom_hline(yintercept = 2,
linetype = "dashed")
scale_y_continuous(limits = c(0,
maximum_y),
breaks = seq(0,
maximum_y,
2))
theme(plot.title = element_text(size = 10,
hjust = 0.5,
face = "bold"),
axis.text.x = element_text(size = 5,
angle = 90,
vjust = 0.5,
hjust = 1),
strip.background = element_rect(fill = "white"),
panel.border = element_rect(colour = "black",
fill = NA),
axis.title.x = element_text(size = 8),
axis.title.y = element_text(size = 8),
plot.background = element_rect(fill = "transparent",
color = NA))
labs(x = "ID",
y = "Log-fold change",
title = "Log-fold change vs sequence")
scale_size(range = c(0.1,
1))
dput(head(my_data_clean_aug_pooling, 30))
structure(list(sample = c("BC372", "BC372", "BC372",
"BC372", "BC372", "BC372", "BC372", "BC372", "BC372", "BC372",
"BC372", "BC372", "BC372", "BC372", "BC372", "BC372", "BC372",
"BC372", "BC372", "BC372", "BC372", "BC372", "BC372", "BC372",
"BC372", "BC372", "BC372", "BC372", "BC372", "BC372"), log_fold_change = c(0.878480955476892,
0.0254158993036971, 0.169690374849339, 1.29365346670481, 0.950207146498172,
0.121582483746693, 0.29591552217522, 0.0493694708020405, 0.253196235065184,
0.511413610788978, 0.92777679529061, 0.633288220541381, 0.852617925189971,
0.245947820840199, 0.284143920808481, 0.54421651055215, 0.998865269852439,
0.468714806763581, 0.704136952532169, 0.334881411284732, 1.09989649348867,
0.44520995356178, 0.559300342753859, 0.198650181166743, 0.947415942094208,
0.0365273151532468, 0.129416762542994, 3.85327690599736, 0.912242173799338,
0.980016944958404), p = c(0.455815003793973, 0.9710277325421,
0.929138758106761, 0.106508575848957, 0.325186030411862, 0.933801784951691,
0.929138758106761, 0.96549305958931, 0.929138758106761, 0.776892782297412,
0.325186030411862, 0.635666815285353, 0.382558882746048, 0.929138758106761,
0.929138758106761, 0.722931599232632, 0.325186030411862, 0.815874297477519,
0.529382980477629, 0.929138758106761, 0.238130758200615, 0.827129935665299,
0.711217028978768, 0.929138758106761, 0.325186030411862, 0.96549305958931,
0.933280410383701, 2.15547277536054e-13, 0.349668295725122, 0.325186030411862
), HLA = c("A0201", "A0201", "A0201", "A0201", "A0201", "A0201",
"A0201", "A0201", "A0201", "A0201", "A0201", "A0201", "A0201",
"A0201", "A0201", "A0201", "A0201", "A0301", "A0301", "A0301",
"A2402", "A2402", "A2402", "A2402", "B0702", "B0702", "B0702",
"B0801", "B0801", "B0801"), Origin = structure(c(5L, 1L, 5L,
9L, 19L, 5L, 7L, 18L, 1L, 3L, 5L, 14L, 14L, 9L, 5L, 3L, 5L, 5L,
5L, 3L, 3L, 2L, 14L, 5L, 3L, 15L, 5L, 5L, 3L, 3L), .Label = c("B19",
"BKPyV", "CMV", "Covid-19", "EBV", "FLU-A", "HAdV-C", "hCoV",
"HHV-1", "HHV-2", "HHV-6B", "HIV-1", "HMPV", "HPV", "JCPyV",
"NWV", "unknown", "VACV", "VZV"), class = "factor"), Peptide = c("v16",
"a47", "a49", "a50", "a51", "a52", "a53", "a55", "a57", "a58",
"a59", "a60", "a61", "a64", "a65", "a66", "a67", "v18", "v25",
"a68", "a74", "a77", "a80", "a81", "v14", "a87", "a89", "v17",
"v22", "a90"), newID = structure(c(3L, 5L, 3L, 5L,
5L, 3L, 5L, 5L, 5L, 1L, 3L, 5L, 5L, 5L, 3L, 1L, 3L, 3L, 3L, 1L,
1L, 5L, 5L, 3L, 1L, 5L, 3L, 3L, 1L, 1L), .Label = c("CMV", "Covid-19",
"EBV", "hCoV", "Other"), class = "factor"), value = c(0, 0, 0,
0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,
0, 0, 0, 1, 0, 0)), row.names = c(NA, -30L), class = c("tbl_df",
"tbl", "data.frame"))
CodePudding user response:
Here is a way.
Instead of filtering the data into 3 different data sets, create a new column with values corresponding to the several conditions. I have called the new variable Colour but any name that makes sense will do. The values are assigned with a case_when statement, where the default is "black". The colours are then manually given in a scale.
To remove the value legend, it now uses argument guide.
I have also defined a custom theme, in order to make the problem code clearer.
theme_LasseVoss <- function(){
theme_minimal() % replace%
theme(
plot.title = element_text(size = 10,
hjust = 0.5,
face = "bold"),
axis.text.x = element_text(size = 5,
angle = 90,
vjust = 0.5,
hjust = 1),
strip.background = element_rect(fill = "white"),
panel.border = element_rect(colour = "black",
fill = NA),
axis.title.x = element_text(size = 8),
axis.title.y = element_text(size = 8),
plot.background = element_rect(fill = "transparent",
color = NA)
)
}
maximum_y <- ceiling(max(my_data_clean_aug_pooling$log_fold_change))
my_data_clean_aug_pooling %>%
mutate(Colour = case_when(
0.001 >= p & log_fold_change >= 2 ~ "interest",
0.001 >= p & log_fold_change < 2 ~ "psig",
TRUE ~ "other"
)) %>%
ggplot(aes(x = Peptide, y = log_fold_change))
geom_point(aes(size = value, colour = Colour))
geom_hline(yintercept = 2, linetype = "dashed")
#
scale_color_manual(
name = "Colour",
values = c(other = "black", interest = "red", psig = "blue")
)
scale_size(
range = c(0.1, 1),
guide = "none"
)
scale_y_continuous(
limits = c(0, maximum_y),
breaks = seq(0, maximum_y, 2)
)
#
labs(x = "ID", y = "Log-fold change",
title = "Log-fold change vs sequence")
facet_grid(.~newID, scales = "free_x", space = "free")
theme_LasseVoss()
CodePudding user response:
Usually it's easier to get the legend working well if you avoid plotting subsets of the data separately, so one way to get your plot is to add an extra column indicating which type of point it is. Note that the example data doesn't include and significant points. You could then change your code to:
my_data_clean_aug_pooling <- my_data_clean_aug_pooling %>%
mutate(group = factor(ifelse(0.001 >= p, ifelse(log_fold_change >= 2, "Interesting", "Significant"), "Boring"),
levels=c("Boring", "Interesting", "Significant")))
levels(my_data_clean_aug_pooling$group)
#[1] "Boring" "Interesting" "Significant"
my_data_clean_aug_pooling %>%
ggplot(aes(x = Peptide,
y = log_fold_change, colour=group, size=group)
)
facet_grid(.~newID,
scales = "free_x",
space = "free")
geom_point()
geom_hline(yintercept = 2,
linetype = "dashed")
scale_y_continuous(limits = c(0,
maximum_y),
breaks = seq(0,
maximum_y,
2))
theme(plot.title = element_text(size = 10,
hjust = 0.5,
face = "bold"),
axis.text.x = element_text(size = 5,
angle = 90,
vjust = 0.5,
hjust = 1),
strip.background = element_rect(fill = "white"),
panel.border = element_rect(colour = "black",
fill = NA),
axis.title.x = element_text(size = 8),
axis.title.y = element_text(size = 8),
plot.background = element_rect(fill = "transparent",
color = NA))
labs(x = "ID",
y = "Log-fold change",
title = "Log-fold change vs sequence")
scale_colour_manual(values=c("Boring"="black","Interesting"="red","Significant"="blue"))
scale_size_manual(values=c("Boring"=0.1, "Interesting"=5, "Significant"=5))
Which gives:
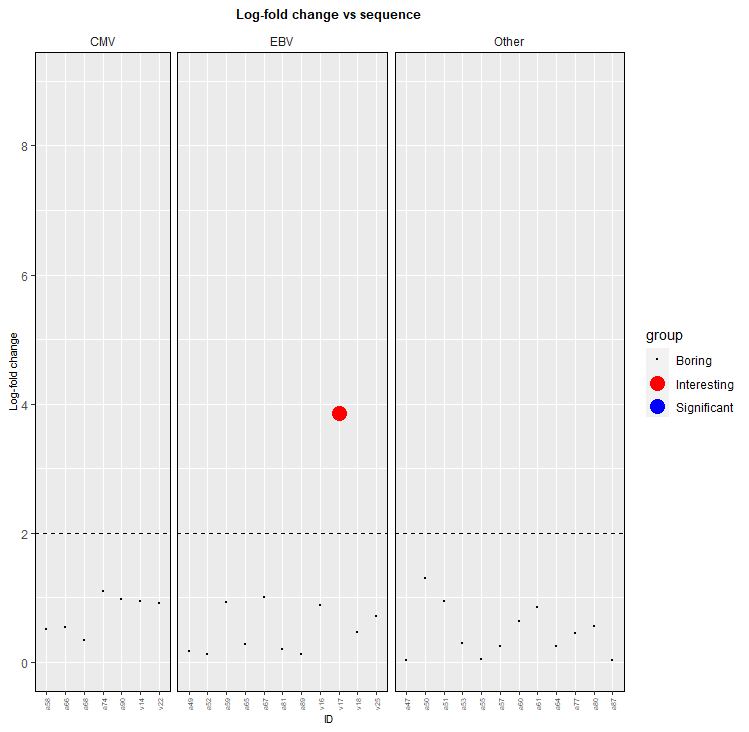
(I may not have chosen the best name for the black points!)