I'm trying to make a single gene coverage plot like the one shown here.
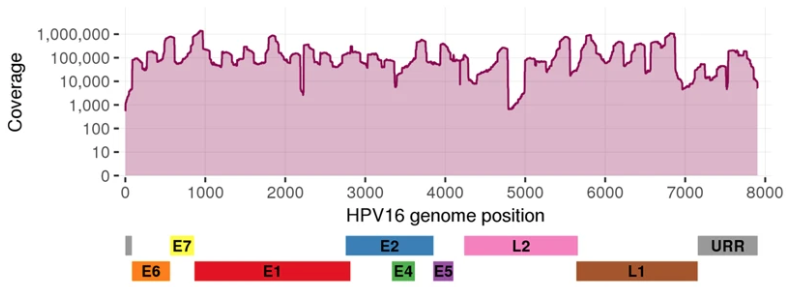
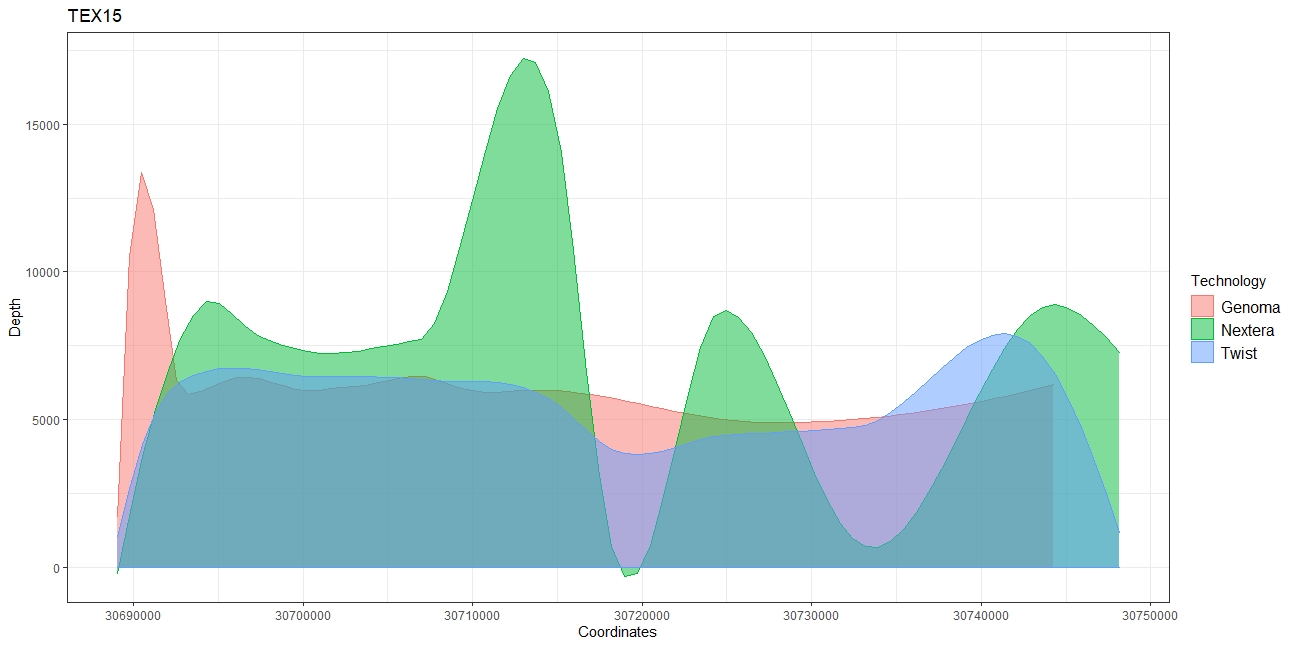
I have already plotted the coverage but I still need to insert the exons and introns in the corresponding coordinates. Thank you! My code:
ggplot(z, aes(x=inicio, y=promedio, fill=Technology, group=Technology, color=Technology))
stat_smooth(
geom = 'area', method = 'loess', span = 1/3,
alpha = 1/2)
scale_x_continuous(limits=c(30689060,30748122))
theme_set(theme_bw())
theme(legend.text = element_text (size = 12))
xlab("Coordinates")
ylab("Depth")
ggtitle("TEX15")
CodePudding user response:
Without a minimal reproducible example (MRE) containing some input data and expected output it's difficult to know how to help you. For instance, here is an MRE with some 'fake' data:
library(tidyverse)
df <- data.frame(Coverage = runif(1000, 0, 7900))
p1 <- df %>%
ggplot(aes(x = Coverage))
geom_density(outline.type = "full", fill = "#D6B7C9")
theme_minimal(base_size = 14)
theme(axis.title.x = element_blank())
features1 <- tribble(~"feature", ~"start", ~"end",
"E1", 1, 1950,
"E5", 2986, 3237,
"L1", 4775, 6292)
features2 <- tribble(~"feature", ~"start", ~"end",
"E2", 1892, 2989,
"L2", 3373, 4794,
"E6", 7125, 7601,
"E7", 7604, 7900)
p2 <- features1 %>%
ggplot()
geom_rect(aes(xmin = start, xmax = end,
ymin = 0, ymax = 1,
fill = feature),
color = "black")
geom_text(aes(x = (start end) / 2, y = 0.5, label = feature))
theme_void()
theme(legend.position = "none")
p3 <- features2 %>%
ggplot()
geom_rect(aes(xmin = start, xmax = end,
ymin = 0, ymax = 1,
fill = feature),
color = "black")
geom_text(aes(x = (start end) / 2, y = 0.5, label = feature))
theme_void()
theme(legend.position = "none")
library(patchwork)
p1 / p2 / p3 plot_layout(nrow = 3, heights = c(1, 0.1, 0.1))
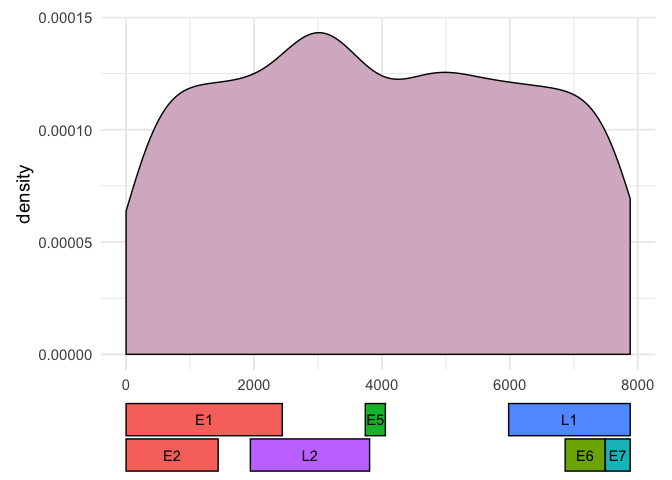
Created on 2022-06-16 by the reprex package (v2.0.1)
Would this approach be suitable for your single gene coverage plot? If not, what would you want to change?