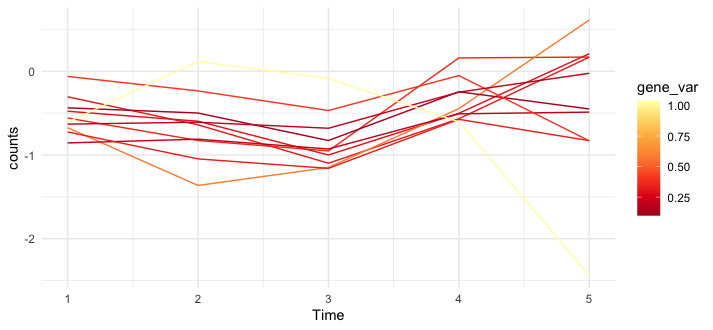
I have clustered my time-series gene expression data based on their temporal expression. Given that there is some variation in the temporal trends, I would like to visualize where most of the lines are to get a more evident trend. One way that I was thinking to do it is to color the lines based on how close they are to the mean trend of the cluster but I have no idea how to implement that.
A reproducible example of my data:
dat <- structure(list(gene_name = c("NIPAL3", "NIPAL3", "NIPAL3", "NIPAL3",
"NIPAL3", "SEMA3F", "SEMA3F", "SEMA3F", "SEMA3F", "SEMA3F", "AOC1",
"AOC1", "AOC1", "AOC1", "AOC1", "ZMYND10", "ZMYND10", "ZMYND10",
"ZMYND10", "ZMYND10", "ACSM3", "ACSM3", "ACSM3", "ACSM3", "ACSM3",
"PDK2", "PDK2", "PDK2", "PDK2", "PDK2", "TMEM132A", "TMEM132A",
"TMEM132A", "TMEM132A", "TMEM132A", "ALDH3B1", "ALDH3B1", "ALDH3B1",
"ALDH3B1", "ALDH3B1", "USH1C", "USH1C", "USH1C", "USH1C", "USH1C",
"NOS2", "NOS2", "NOS2", "NOS2", "NOS2"), Sample.name = c("DMSO_1",
"DMSO_2", "DMSO_24", "DMSO_4", "DMSO_8", "DMSO_1", "DMSO_2",
"DMSO_24", "DMSO_4", "DMSO_8", "DMSO_1", "DMSO_2", "DMSO_24",
"DMSO_4", "DMSO_8", "DMSO_1", "DMSO_2", "DMSO_24", "DMSO_4",
"DMSO_8", "DMSO_1", "DMSO_2", "DMSO_24", "DMSO_4", "DMSO_8",
"DMSO_1", "DMSO_2", "DMSO_24", "DMSO_4", "DMSO_8", "DMSO_1",
"DMSO_2", "DMSO_24", "DMSO_4", "DMSO_8", "DMSO_1", "DMSO_2",
"DMSO_24", "DMSO_4", "DMSO_8", "DMSO_1", "DMSO_2", "DMSO_24",
"DMSO_4", "DMSO_8", "DMSO_1", "DMSO_2", "DMSO_24", "DMSO_4",
"DMSO_8"), Treatment = c("DMSO", "DMSO", "DMSO", "DMSO", "DMSO",
"DMSO", "DMSO", "DMSO", "DMSO", "DMSO", "DMSO", "DMSO", "DMSO",
"DMSO", "DMSO", "DMSO", "DMSO", "DMSO", "DMSO", "DMSO", "DMSO",
"DMSO", "DMSO", "DMSO", "DMSO", "DMSO", "DMSO", "DMSO", "DMSO",
"DMSO", "DMSO", "DMSO", "DMSO", "DMSO", "DMSO", "DMSO", "DMSO",
"DMSO", "DMSO", "DMSO", "DMSO", "DMSO", "DMSO", "DMSO", "DMSO",
"DMSO", "DMSO", "DMSO", "DMSO", "DMSO"), Time = c(1, 2, 5, 3,
4, 1, 2, 5, 3, 4, 1, 2, 5, 3, 4, 1, 2, 5, 3, 4, 1, 2, 5, 3, 4,
1, 2, 5, 3, 4, 1, 2, 5, 3, 4, 1, 2, 5, 3, 4, 1, 2, 5, 3, 4, 1,
2, 5, 3, 4), counts = c(-0.674728025769495, -1.36397354466049,
0.610379522258895, -1.14928773078363, -0.446571694189183, -0.436985550549815,
-0.499130283673904, -0.450641960945776, -0.827458887288972, -0.245426299345142,
-0.305285624771504, -0.640229241821068, 0.167251080770738, -1.09858244762228,
-0.567447650338103, -0.609472980799307, 0.115975465710009, -2.43581298419879,
-0.085029879459607, -0.604272206569522, -0.559054804869209, -0.827020967548349,
0.168793788002722, -0.951210549139716, 0.15852569469711, -0.724991719902859,
-1.04664568307064, -0.828798242883774, -1.15973081519968, -0.574087099835571,
-0.0620293805158624, -0.235285085548273, -0.830591518382229,
-0.469245592463803, -0.0521555714928603, -0.477982049612678,
-0.59701614315213, 0.208614404395684, -1.00028569852069, -0.503026174159395,
-0.631726558325577, -0.60884838805659, -0.0250059525913151, -0.680997114400299,
-0.249988532737743, -0.856318481580249, -0.811356236427306, -0.48845066252092,
-0.928288203615435, -0.508993568295652)), row.names = c(NA, -50L
), class = c("tbl_df", "tbl", "data.frame"))
Gene_name column contains the genes that I have used for clustering. Sample name contains the biological conditions the measurements were taken and the time. E.g DMSO_1 corresponds to the measurement in DMSO samples at 1h. The same information are found also in two separate columns (i.e. Treatment and Time) The counts column correspond to the mean, scaled counts of a gene, and the cluster column is the cluster that each gene was assigned (in total I have 8 clusters).
I am plotting my data using
dat_r%>%
ggplot(aes(Time, counts))
geom_line(aes(group = gene_name, alpha = 0.3))
geom_smooth(stat = "summary", fun = "mean")
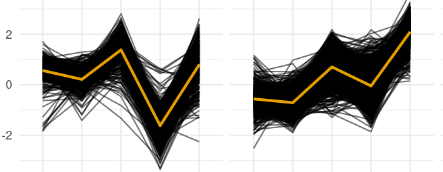
An example of a cluster I am getting using the code above with my complete data.
and an example of the desired output is (taken from 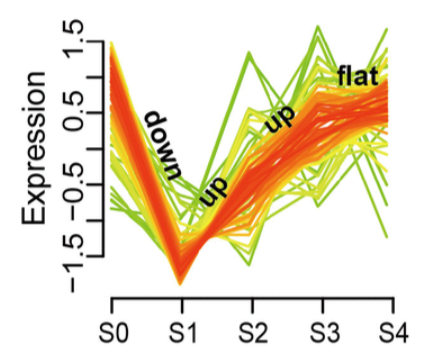
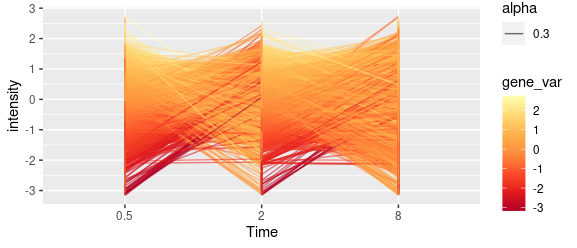
I have tried to use the following code to color each line based on its distance from the mean trend of a cluster.
dat %>%
group_by(gene_name) %>%
mutate(gene_var = counts - mean(counts, na.rm = TRUE)) %>%
ggplot(aes(Time,counts, color = gene_var))
geom_line(aes(group = gene_name, alpha = 0.3))
scale_color_distiller(palette = "YlOrRd")
but the returning plot is not coloring the line with one color.
CodePudding user response:
Here's a go using the storms data from dplyr. I calculate each storms RMSE vs the average windspeed trajectory. Maybe not the best example but I think it does what you want.
# make some example data
storms_sample <- storms %>%
group_by(name, year) %>%
transmute(wind, obs = row_number()) %>%
filter(obs <= 60, max(obs) >= 55)
# calculate RMSE for each storm, and plot based on that
storms_sample %>%
group_by(obs) %>%
mutate(obs_var = wind - mean(wind, na.rm = TRUE)) %>%
group_by(name, year) %>%
mutate(storm_var = sqrt(mean(obs_var^2))) %>%
ungroup() %>% # EDIT - might fix OP's problem
filter(!storm_var %>% between(15,30)) %>% # Edit to highlight extremes
ggplot(aes(obs, wind, group = interaction(name,year), color = storm_var))
geom_line(alpha = 0.6)
scale_color_distiller(palette = "YlOrRd")
theme_minimal()
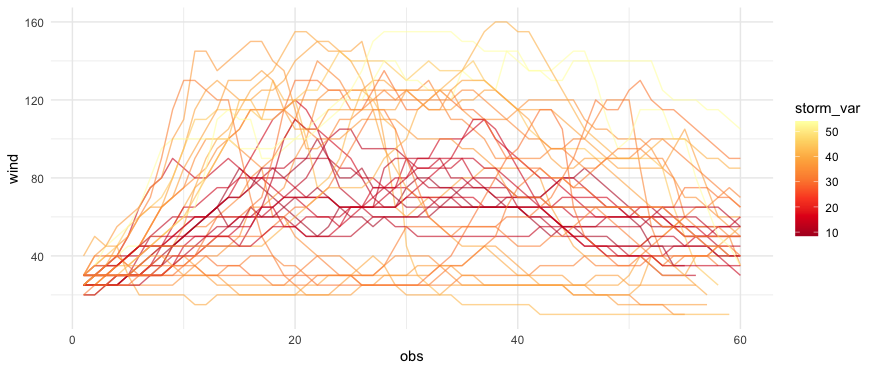
EDIT - here's code I tried on the new sample data, which seems to work -- e.g. there's a gene_var that doesn't follow the trend which is much more yellow than the others.
dat %>%
group_by(Time) %>%
mutate(var = counts - mean(counts, na.rm = TRUE)) %>%
group_by(gene_name) %>%
mutate(gene_var = sqrt(mean(var^2))) %>%
ungroup() %>%
ggplot(aes(Time, counts, group = gene_name, color = gene_var))
geom_line()
scale_color_distiller(palette = "YlOrRd")
theme_minimal()