I have dataset from my analysis. To interpretate the result, I am trying to build a dataframe
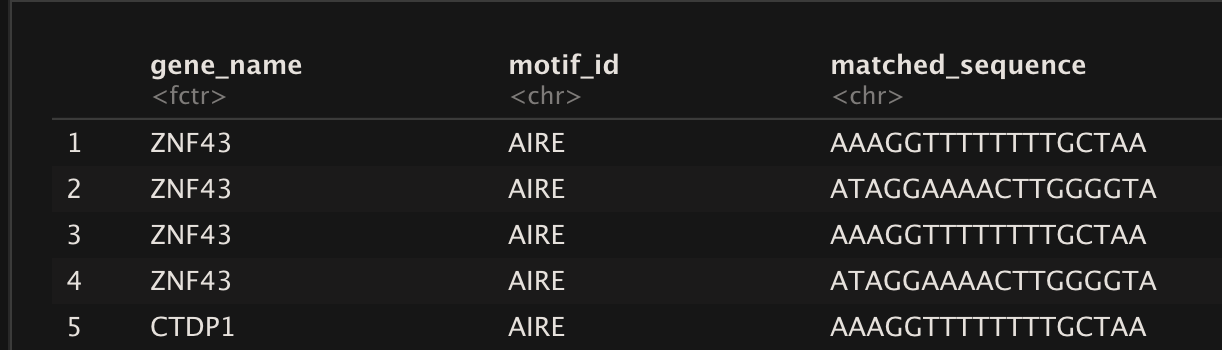
Result should be like :
gene_name | Motif_id_1 | Motif_id_2 | Occurence | Matched_sequence

here some motif_id may share gene_name and result should be two combination of motif_id(overlap allowed.)
I have tried following code, however the result does not give combination within motif_id.
merge_practice <- reshape2::dcast(group_geneid_CT,
motif_id motif_id~gene_name,
value.var ="matched_sequence",
drop = T,fill = 0,
fun.aggregate = length )
If possible, I want to make it memory and time efficient and less dependency with packages. Can anyone give me an another perspective?
CodePudding user response:
library(tidyverse)
data <- tribble(
~gene_name, ~motif_id, ~matched_sequence,
"A", "y1", "ccc",
"A", "y2", "ccc",
"A", "y1", "aaa",
"A", "y2", "aaa",
"A", "y2", "aat",
)
data %>%
pull(motif_id) %>%
unique() %>%
combn(2) %>%
t() %>%
as_tibble() %>%
rename(from = V1, to = V2) %>%
mutate(
co_occurrence = list(from, to) %>% pmap(~ {
bind_rows(
data %>% filter(motif_id == .x) %>% select(-motif_id),
data %>% filter(motif_id == .y) %>% select(-motif_id)
) %>%
count(gene_name, matched_sequence, name = "co_occurrent")
})
) %>%
unnest(co_occurrence)
#> Warning: The `x` argument of `as_tibble.matrix()` must have unique column names if `.name_repair` is omitted as of tibble 2.0.0.
#> Using compatibility `.name_repair`.
#> # A tibble: 3 × 5
#> from to gene_name matched_sequence co_occurrent
#> <chr> <chr> <chr> <chr> <int>
#> 1 y1 y2 A aaa 2
#> 2 y1 y2 A aat 1
#> 3 y1 y2 A ccc 2
Created on 2022-03-01 by the reprex package (v2.0.0)
co_occurrent should be either 2 if it was found in both motifs or 1 if it was only found in one motif.