I can see many posts on this topic, but none addresses this question. Apologies if I missed a relevant answer. I have a large protein expression dataset, with samples like so as the columns: rep1_0hr, rep1_16hr, rep1_24hr, rep1_48hr, rep1_72hr .....
and 2000 proteins in the rows. In other words each sample is a different developmental timepoint.
If it is of any interest, the original dataset is 'mulvey2015' from the pRolocdata package in R, which I converted to a SummarizedExperiment object in RStudio.
I first ran k-means clustering on the data (an assay() of a SummarizedExperiment dataset, to get 12 clusters:
k_mul <- kmeans(scale(assay(mul)), centers = 12, nstart = 10)
Then:
summary(k_mul)
produced the expected output.
I would like the visualisation to look like this, with samples on the x-axis and expression on the y-axis. The plots look like they have been generated using facet_wrap() in ggplot:
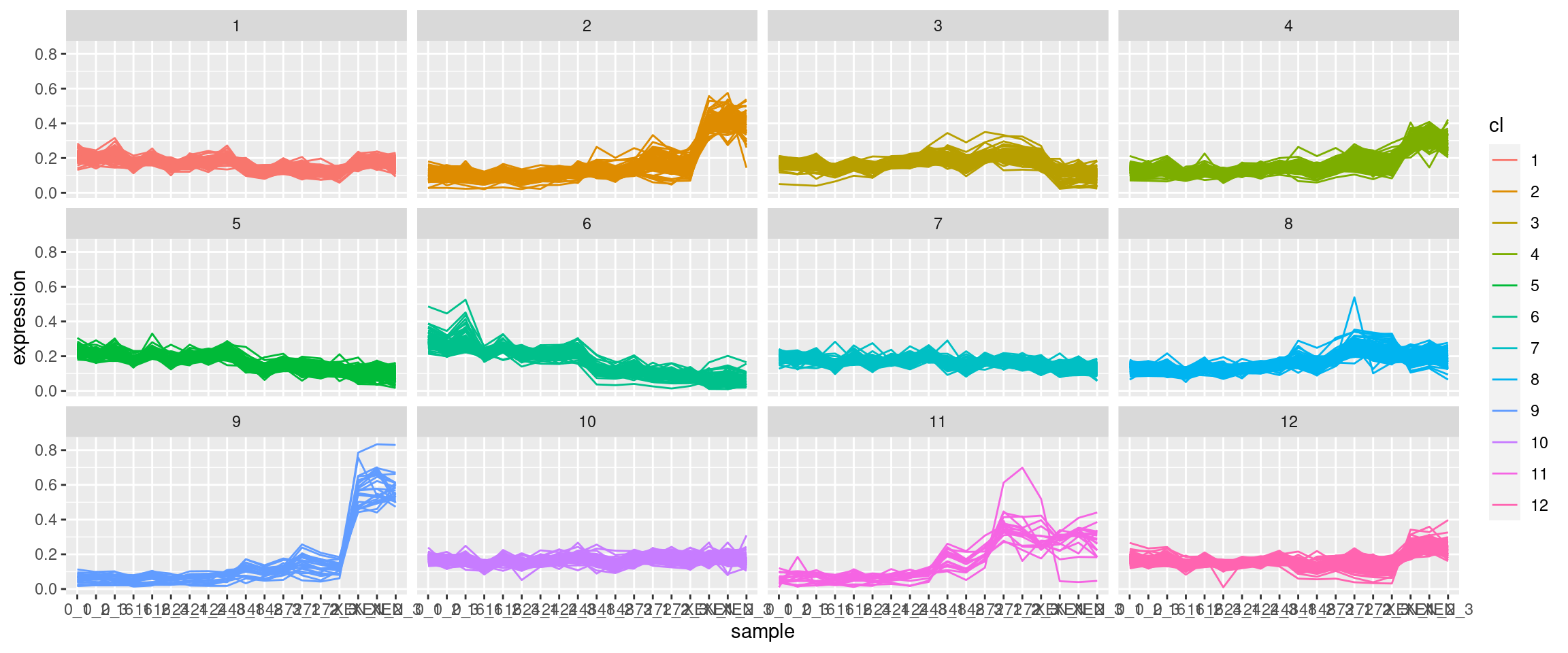
For ggplot the data need to be provided as a dataframe with a column for the cluster identity of an individual protein. Also the data need to be in long format. I tried pivoting (pivot_longer) the original dataset, but of course there are a very large number of data points. Moreover, the image I posted shows that for any one plot, the number of coloured lines is smaller than the total number of proteins, suggesting that there might have been dimension reduction on the dataset first, but I am unsure. Up till now I have been running the kmeans algorithm without dimension reduction. Can I get guidance please for how to produce this plot?
CodePudding user response:
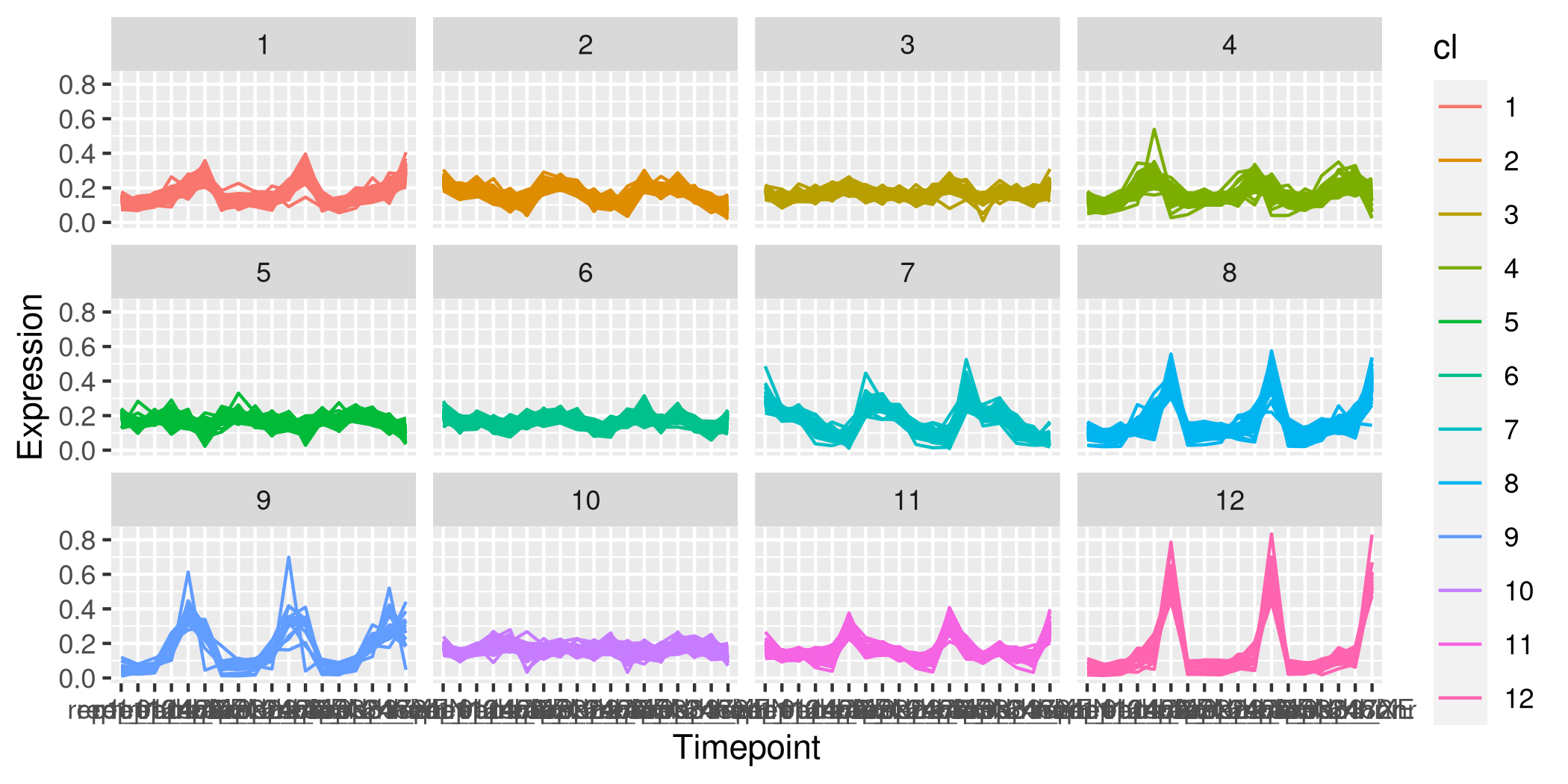
Here is my attempt at reverse engeneering the plot:
library(pRolocdata)
library(dplyr)
library(tidyverse)
library(magrittr)
library(ggplot2)
mulvey2015 %>%
Biobase::assayData() %>%
magrittr::extract2("exprs") %>%
data.frame(check.names = FALSE) %>%
tibble::rownames_to_column("prot_id") %>%
mutate(.,
cl = kmeans(select(., -prot_id),
centers = 12,
nstart = 10) %>%
magrittr::extract2("cluster") %>%
as.factor()) %>%
pivot_longer(cols = !c(prot_id, cl),
names_to = "Timepoint",
values_to = "Expression") %>%
ggplot(aes(x = Timepoint, y = Expression, color = cl))
geom_line(aes(group = prot_id))
facet_wrap(~ cl, ncol = 4)
As for you questions, pivot_longer is usually quite performant unless it fails to find unique combinations in keys or problems related with data type conversion. The plot can be improved by:
- tweaking the
alphaparameter ofgeom_lines(e.g. alpha = 0.5), in order to provide an idea of density of lines - finding a good abbreviation and order for
Timepoint - changing axis.text.x orientation
CodePudding user response:
Here is my own, very similar solution to the above.
dfsa_mul <- data.frame(scale(assay(mul)))
dfsa_mul2 <- rownames_to_column(dfsa_mul, "protID")
add the kmeans $cluster column to the dfsa_mul2 dataframe. Only change clus to a factor after executing pivot_longer
dfsa_mul2$clus <- ksa_mul$cluster
dfsa_mul2 %>%
pivot_longer(cols = -c("protID", "clus"),
names_to = "samples",
values_to = "expression") %>%
ggplot(aes(x = samples, y = expression, colour = factor(clus)))
geom_line(aes(group = protID))
facet_wrap(~ factor(clus))
This generates a series of plots identical to the graphs posted by @sbarbit.